Neonatal Hypotonia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Schematic Approach to Hypotonia in Infancy
Leyenaar.qxd 8/26/2005 4:03 PM Page 397 NEUROLOGY SUBSPECIALTY ARTICLE A schematic approach to hypotonia in infancy JoAnna Leyenaar MD MPH, Peter Camfield MD FRCPC, Carol Camfield MD FRCPC J Leyenaar, P Camfield, C Camfield. A schematic approach Une démarche schématique envers l’hypotonie to hypotonia in infancy. Paediatr Child Health 2005; pendant la première enfance 10(7):397-400. L’hypotonie peut être le signe révélateur de nombreuses maladies Hypotonia may be the presenting sign for many systemic diseases and systémiques ou du système nerveux. Le présent article traite d’une diseases of the nervous system. The present paper discusses a rational, démarche diagnostique rationnelle, simple et précise envers l’hypotonie simple and accurate diagnostic approach to hypotonia in infancy, pendant la première enfance, illustrée par le cas d’une fillette de cinq mois illustrated by the case of a five-month-old infant girl recently referred récemment aiguillée vers le IWK Health Centre de Halifax, en Nouvelle- to the IWK Health Centre in Halifax, Nova Scotia. Key points in the Écosse. Les principaux points de l’anamnèse et de l’examen physique sont history and physical examination are outlined to allow a tailored exposés afin de permettre une exploration personnalisée de la patiente et investigation both for the patient and for other hypotonic infants. A des autres nourrissons hypotoniques. Un exposé sur une importante discussion of an important neuromuscular disease, diagnosed in the maladie neuromusculaire, diagnostiquée chez la patiente, conclut l’article. present patient, concludes the paper. Key Words: Hypotonia; Infant; Spinal muscular atrophy nfants with hypotonia pose challenges for clinicians respiratory syncytial virus-positive bronchiolitis. -

Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H
Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H. Long, MD, PhD,a,b Jennifer G. Fiore, MD,a,b Riaz Gillani, MD,a,b Laurie M. Douglass, MD,c Alan M. Fujii, MD,d Jodi D. Hoffman, MDe A 2-day old term male infant was found to be hypotonic and minimally abstract reactive during routine nursing care in the newborn nursery. At 40 hours of life, he was hypoglycemic and had intermittent desaturations to 70%. His mother had an unremarkable pregnancy and spontaneous vaginal delivery. The mother’s prenatal serology results were negative for infectious risk factors. Apgar scores were 9 at 1 and 5 minutes of life. On day 1 of life, he fed, stooled, and voided well. Our expert panel discusses the differential diagnosis of hypotonia in a neonate, offers diagnostic and management recommendations, and discusses the final diagnosis. DRS LONG, FIORE, AND GILLANI, birth weight was 3.4 kg (56th PEDIATRIC RESIDENTS percentile), length was 52 cm (87th aDepartment of Medicine, Boston Children’s Hospital, d e percentile), and head circumference Boston, Massachusetts; and Neonatology Section, Medical A 2-day old male infant born at Genetics Section, cDivision of Child Neurology, and 38 weeks and 4 days was found to be was 33 cm (12th percentile). His bDepartment of Pediatrics, Boston Medical Center, Boston, limp and minimally reactive during physical examination at birth was Massachusetts routine care in the newborn nursery. normal for gestational age, with Drs Long, Fiore, and Gillani conceptualized, drafted, Just 5 hours before, he had an appropriate neurologic, cardiac, and and edited the manuscript; Drs Douglass, Fujii, and appropriate neurologic status when respiratory components. -

Hypotonia Surestep Product Catalog Page 29 in Step with Pediatric Hypotonia
SPECIAL EDUCATIONAL SERIES DIAGNOSTIC INSIGHTS ANALYZING GAIT CHANGES GROSS MOTOR SKILLS ORTHOTIC MANAGEMENT CLI N I CAL CASE STUDIES Sponsored by an educational grant from: In Step With Pediatric Hypotonia SureStep Product Catalog Page 29 In Step With Pediatric Hypotonia Contents VIEWPOINT FROM THE EDITOR: An Unexpected Path, Mobility and More an Invaluable Perspective At the most basic level, mobility is about get- PAGE 3 ting from point A to point B. But, for many children with hypotonia, it’s about so much 4 more. FEATURES It’s about independence. It’s about con- fidence. It’s about maintaining strength, fit- ness, and healthy bones. It’s about not being Understanding Hypotonia excluded from activities enjoyed by their PAGE 4 typically developing peers. And improved mobility may have even Gait: The Cornerstone more benefits in those children whose hy- potonia is associated with social and behav- of Intervention ioral developmental delays. New research PAGE 8 has identified an association between motor skills and sociobehavioral milestones in chil- 8 The Importance of Gross dren with autism spectrum disorder, who often present with hypotonia (see “The Im- Motor Skills portance of Gross Motor Skills,” page 12). PAGE 12 This suggests that early intervention to improve gross motor skills—including or- thotic devices and physical therapy—may Orthotic Solutions for also help certain children interact more Children with Hypotonia comfortably with others. That won’t come as PAGE 16 a surprise to the clinicians and parents who 12 have personally seen it happen. This special issue is filled with evidence- Orthotic Success Stories: based information and personal success sto- Four Cases in a Series ries illustrating how effective interventions can enhance mobility in children with hy- PAGE 20 potonia. -

Myasthenia and Related Disorders of the Neuromuscular Junction Jennifer Spillane, David J Beeson, Dimitri M Kullmann
Myasthenia and related disorders of the neuromuscular junction Jennifer Spillane, David J Beeson, Dimitri M Kullmann To cite this version: Jennifer Spillane, David J Beeson, Dimitri M Kullmann. Myasthenia and related disorders of the neuromuscular junction. Journal of Neurology, Neurosurgery and Psychiatry, BMJ Publishing Group, 2010, 81 (8), pp.850. 10.1136/jnnp.2008.169367. hal-00557404 HAL Id: hal-00557404 https://hal.archives-ouvertes.fr/hal-00557404 Submitted on 19 Jan 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Myasthenia and related disorders of the neuromuscular junction Jennifer Spillane1, David J Beeson2 and Dimitri M Kullmann1 1UCL Institute of Neurology 2Weatherall Institute for Molecular Medicine, Oxford University Abtract Our understanding of transmission at the neuromuscular junction has increased greatly in recent years. We now recognise a wide variety of autoimmune and genetic diseases that affect this specialised synapse, causing muscle weakness and fatigue. These disorders greatly affect quality of life and rarely can be fatal. Myasthenia Gravis is the most common disorder and is most commonly caused by auto‐antibodies targeting postsynaptic acetylcholine receptors (AChRs). Antibodies to muscle‐specific kinase (MuSK) are detected in a variable proportion of the remainder. -

Combined Web 759..782
Movement Disorders Vol. 24, No. 5, 2009, pp. 759–782 Ó 2009 Movement Disorder Society Brief Reports Clinical Characteristics of Psychogenic movement disorders (PMDs) are not uncommon in movement disorder clinics.1 PMDs may 49 Patients with Psychogenic phenomenologically mimic almost all movement disor- Movement Disorders in a Tertiary ders. The most common movement disorder is tremor, followed by dystonia and others.2–5 Clinic in Turkey Diagnostic criteria for PMDs was first identified by Fahn and Williams, based on atypical and common Sibel Ertan, MD,1 Derya Uluduz, MD,1 clinical clues.6 Later, other authors described additional 1* 1 Sibel O¨ zekmekc¸i, MD, Gu¨nes Kiziltan, MD, features to distinguish PMD patients from those with 2 1 1 Turan Ertan, MD, Cengiz Yalc¸inkaya, MD, , and neurogenic movement disorders.7–9 ¨ 1 and C¸ igdem Ozkara, MD Because there is no study written in English on any 1Department of Neurology, Cerrahpasa Faculty of Medicine, hospital-based data of PMDs in Turkey, we aimed to Istanbul University, Istanbul, Turkey; 2Department of identify the frequency and phenomenological features Psychiatry, Cerrahpasa Faculty of Medicine, Istanbul of PMDs in our patient population with movement dis- University, Istanbul, Turkey orders. Abstract: Patients admitted to movement disorders outpa- tient unit at a university hospital between January 2002 and June 2007 were screened for psychogenic movement PATIENTS AND METHODS disorders (PMDs). Out of 1,743 patients, 49 patients Patients admitted to our Movement Disorders Unit (2.8%), including four children, were diagnosed to have between January 2002 and June 2007, were screened PMDs. Women to men ratio was 34/15. -
IUGR: Intrauterine Growth Restriction
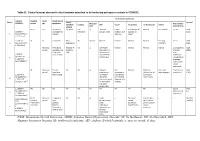
Table S1. Clinical features observed in the 6 patients described so far harboring pathogenic variants in FOXRED1. Evolutionary symptoms Variants Prenatal Onset Onset clinical Patient Lactic/ Survival FOXRED1 period age symptoms Muscular Psychomotor Metabolic Epilepsy MRI Visual Respiratory Cardiovascular Others tone development acidosis IUGR 2m Hypotonia Yes (+++) Yes ↓ Normal Latent Bronchospasm Normal AEP normal IQ: 48 Alive c.920G>A Development refractary (2m,4y,7y3m) strabismus of episodes in (15y) 1 (p.Gly307Glu) / al delay right eye infant c.733+1G>A c.920G>A NI 4y Clumsiness With No Normal Normal Normal Normal Normal Learning IQ: 99 Alive 2 (p.Gly307Glu) / exercise (+) disorders (19y) c.733+1G>A - Neonatal Premature; No (only ↑ Yes ↓ Decreased Normal Normal Normal Normal Gradually loss Alive period Hypoglycemia lactate in attenuation in of motor (22y)) Congenital LCR) the putamen skills; c.694C>T lactic acidosis and cerebellar wheelchair; (p.Gln232X) / 3 atrophy (6y) no expressive c.1289A>G language; 18 (p.Asn430Ser) understands simple commands NI Neonatal Truncal Yes Yes ↓ Delayed Eye Normal Mild non- Persistent Psychomotor Alive period hypotonia myelination movements obstructive left hepatomegaly retardation (10y) c.1054 C>T Poor feeding ventricular have always ventricular (p.Arg352Trp) / dilatation; been roving hypertrophy 4 c.1054 C>T abnormal signal bilateral optic (p.Arg352Trp) 19 in the thalami atrophy and basal ganglia (8m) c.1308G>A ND ND ND ND Yes NA NA NA NA NA NA Severe Alive (p.Val421Met) / psychomotor (¿) 5 c.1308G>A retardation (p.Val421Met)20 IUGR; Neonatal Congenital Yes Yes ↓ Large -- Persistent Dilated right - - Death (3 c.612_615dupA Cerebral period lactic periventricular severe ventricle and months) CTG intraventric acidosis. -

Cerebral Hypotonia by Mihee Bay MD (Dr
Cerebral hypotonia By Mihee Bay MD (Dr. Bay of Kennedy Krieger Institute and Johns Hopkins School of Medicine has no relevant financial relationships to disclose.) Originally released July 12, 2006; last updated February 1, 2016; expires February 1, 2019 Introduction This article includes discussion of cerebral hypotonia, central hypotonia, essential hypotonia, benign congenital hypotonia, and floppy infant. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations. Overview Hypotonia is a clinical manifestation of numerous diseases affecting the central and/or peripheral motor nervous system. The key to accurate diagnosis involves integral steps of evaluation that include a detailed history, examination, and diagnostic tests. “Cerebral” (or central) hypotonia implies pathogenesis from abnormalities from the central nervous system, and related causal disorders include cerebral dysgenesis and genetic or metabolic disorders. Patients with central hypotonia generally have hypotonia without associated weakness, in contrast to the peripheral (lower motor neuron) causes, which typically produce both hypotonia and muscle weakness. Hypotonia is a clinical manifestation of over 500 genetic disorders; thus, a logical, stepwise approach to diagnosis is essential. With recent advances in the field of genetic testing, diagnostic yield will undoubtedly improve. There is no cure, but treatment includes supportive therapies, such as physical and occupational therapy, and diagnosis-specific management. Key points • Hypotonia is reduced tension or resistance of passive range of motion. • The first step in the evaluation of a child with hypotonia is localization to the central (“cerebral”) or peripheral nervous system, or both. • Central hypotonia is more likely to be noted axially with normal strength and hyperactive to normal deep tendon reflexes. -

Diagnosis and Treatment of Facioscapulohumeral Muscular Dystrophy: 2015 Guidelines Steven Karceski Neurology 2015;85;E41-E43 DOI 10.1212/WNL.0000000000001865
PATIENT PAGE Section Editors Diagnosis and treatment of DavidC.Spencer,MD Steven Karceski, MD facioscapulohumeral muscular dystrophy 2015 guidelines Steven Karceski, MD WHAT DID THE AUTHORS STUDY? Dr. Tawil led a in people with FSHD. However, a person with committee of doctors who specialize in diagnosing FSHD could develop heart problems unrelated to and treating facioscapulohumeral muscular dystrophy FSHD. If a person with FSHD developed heart prob- (FSHD). Together, they reviewed published articles lems, he or she would need to see a doctor for an eval- and research in FSHD and similar muscular dystro- uation and treatment. phies. They assembled detailed recommendations Although rare, patients with a low number of about the diagnosis and treatment of people with copies of D4Z4 may develop problems with their FSHD.1 vision. They develop Coats disease, which can be de- tected by an ophthalmologist using special equip- HOW IS FSHD DIAGNOSED? The initial step to the ment called indirect ophthalmoscopy. In short, a diagnosis of FSHD is taking a careful medical history. person who has a low number of copies should be This starts in the doctor’s office. The doctor will ask screened and evaluated for this possibility by a many questions about the person’s weakness: how it trained eye specialist. started, where it is most noticeable, how quickly it is Pain is common in people with FSHD. The pain worsening, and whether there is a family history of occurs in the muscles and bones. It often responds to the same kind of problem. If there is a family history several medications and physical therapy. -

Tremor in X-Linked Recessive Spinal and Bulbar Muscular Atrophy (Kennedy’S Disease)
CLINICS 2011;66(6):955-957 DOI:10.1590/S1807-59322011000600006 CLINICAL SCIENCE Tremor in X-linked recessive spinal and bulbar muscular atrophy (Kennedy’s disease) Francisco A. Dias,I Renato P. Munhoz,I Salmo Raskin,II Lineu Ce´sar Werneck,I He´lio A. G. TeiveI I Movement Disorders Unit, Neurology Service, Internal Medicine Department, Hospital de Clı´nicas, Federal University of Parana´ , Curitiba, PR, Brazil. II Genetika Laboratory, Curitiba, PR, Brazil. OBJECTIVE: To study tremor in patients with X-linked recessive spinobulbar muscular atrophy or Kennedy’s disease. METHODS: Ten patients (from 7 families) with a genetic diagnosis of Kennedy’s disease were screened for the presence of tremor using a standardized clinical protocol and followed up at a neurology outpatient clinic. All index patients were genotyped and showed an expanded allele in the androgen receptor gene. RESULTS: Mean patient age was 37.6 years and mean number of CAG repeats 47 (44-53). Tremor was present in 8 (80%) patients and was predominantly postural hand tremor. Alcohol responsiveness was detected in 7 (88%) patients with tremor, who all responded well to treatment with a b-blocker (propranolol). CONCLUSION: Tremor is a common feature in patients with Kennedy’s disease and has characteristics similar to those of essential tremor. KEYWORDS: Kennedy’s disease; X-linked recessive bulbospinal neuronopathy; Spinal and bulbar muscular atrophy; Motor neuron disease; Tremor. Dias FA, Munhoz RP, Raskin S, Werneck LC, Teive HAG. Tremor in X-linked recessive spinal and bulbar muscular atrophy (Kennedy’s disease). Clinics. 2011;66(6):955-957. Received for publication on December 24, 2010; First review completed on January 18, 2011; Accepted for publication on February 25, 2011 E-mail: [email protected] Tel.: 55 41 3019-5060 INTRODUCTION compatible with a long life. -

Mean Platelet Volume in Asymptomatic Chorioamnionitis-Exposed Infants
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2021;10(1):e100132 doi: 10.7363/100132 Received: 2019 Aug 22; revised: 2020 Jan 26; accepted: 2020 Feb 02; published online: 2020 Dec 28 Original article Mean platelet volume in asymptomatic chorioamnionitis- exposed infants. A retrospective case-control study Atef Alshafei, Moustafa Hassan, Yaser El saba, Anwar Khan, Mahmoud Ahmed Neonatology Section, Pediatric Department, Dubai Hospital, Dubai, UAE Abstract Introduction: Maternal chorioamnionitis (CA) is a serious condition causing several neonatal morbidities and long-term neurodevelopmental sequelae in exposed infants. Current guidelines still recommend admission, laboratory evaluation, and antibiotic administration to all CA-exposed infants. The incidence of early-onset neonatal sepsis (EOS) is currently low, owing to the routine intrapartum antibiotic administration to mothers identified to be at risk of developing CA. New diagnostic tools for early diagnosis of sepsis in apparently healthy infants exposed to maternal CA are needed. Previous studies showed that mean platelet volume (MPV) is evolving as a potential inflammatory marker of neonatal sepsis. We aimed to study whether MPV can be used as an adjuvant diagnostic tool for EOS in asymptomatic CA-exposed infants. Objective: To evaluate the role of MPV as an adjuvant biomarker of EOS in cases of asymptomatic CA-exposed infants. Design: Retrospective case-control study. Setting: A tertiary care Neonatal Intensive Care Unit (NICU). Patients: Asymptomatic CA-exposed infants 37-40 weeks of gestation admitted between May 2016 and April 2019 to the NICU of Dubai Hospital, UAE. Results: A total of 1,300 infants were admitted to NICU during the study period. -

Consensus-Based Care Recommendations for Adults with Myotonic Dystrophy Type 1
Consensus-based Care Recommendations for Adults with Myotonic Dystrophy Type 1 I Consensus-based Care Recommendations for Adults with Myotonic Dystrophy Type 1 Due to the multisystemic nature of this disease, the studies and rigorous evidence needed to drive the creation of an evidence-based guideline for the clinical care of adult myotonic dystrophy type 1 (DM1) patients are not currently available for all affected body systems and symptoms. In order to improve and standardize care for this disorder now, more than 65 leading myotonic dystrophy (DM) clinicians in Western Europe, the UK, Canada and the US joined in a process started in Spring 2015 and concluded in Spring 2017 to create the Consensus-based Care Recommendations for Adults with Myotonic Dystrophy Type 1. The project was organized and supported by the Myotonic Dystrophy Foundation (MDF). A complete list of authors and an overview of the process is available in Addendum 1. A complete reading list for each of the study area sections is available in Addendum 2. An Update Policy has been adopted for this document and will direct a systematic review of literature and appropriate follow up every three years. Myotonic Dystrophy Foundation staff will provide logistical and staff support for the update process. A Quick Reference Guide extrapolated from the Consensus-based Care Recommendations is available here http://www.myotonic.org/clinical-resources For more information, visit myotonic.org. Myotonic Dystrophy Foundation 1 www.myotonic.org Table of Contents Life-threatening symptoms -

Evidence-Based Guideline: Evaluation, Diagnosis, and Management Of
Evidence-based Guideline: Evaluation, Diagnosis, and Management of Facioscapulohumeral Muscular Dystrophy Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine Rabi Tawil, MD, FAAN1; John T. Kissel, MD, FAAN2; Chad Heatwole, MD, MS-CI3; Shree Pandya, PT, DPT, MS4; Gary Gronseth, MD, FAAN5; Michael Benatar, MBChB, DPhil, FAAN6 (1) MDA Neuromuscular Disease Clinic, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (2) Department of Neurology, Wexner Medical Center, Ohio State University, Columbus, OH (3) Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (4) Department of Neurology, School of Medicine and Dentistry, University of Rochester Medical Center, Rochester, NY (5) Department of Neurology, University of Kansas School of Medicine, Kansas City, KS (6) Department of Neurology, Miller School of Medicine, University of Miami, Miami, FL Correspondence to: American Academy of Neurology [email protected] 1 Approved by the Guideline Development, Dissemination, and Implementation Subcommittee on July 23, 2014; by the AAN Practice Committee on October 20, 2014; by the AANEM Board of Directors on [date]; and by the AANI Board of Directors on [date]. This guideline was endorsed by the FSH Society on December 18, 2014. 2 AUTHOR CONTRIBUTIONS Rabi Tawil: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. John Kissel: acquisition of data, analysis or interpretation of data, critical revision of the manuscript for important intellectual content.