Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management of Late Preterm and Term Neonates Exposed to Maternal Chorioamnionitis Mitali Sahni1,4* , María E
Sahni et al. BMC Pediatrics (2019) 19:282 https://doi.org/10.1186/s12887-019-1650-0 RESEARCH ARTICLE Open Access Management of Late Preterm and Term Neonates exposed to maternal Chorioamnionitis Mitali Sahni1,4* , María E. Franco-Fuenmayor2 and Karen Shattuck3 Abstract Background: Chorioamnionitis is a significant risk factor for early-onset neonatal sepsis. However, empiric antibiotic treatment is unnecessary for most asymptomatic newborns exposed to maternal chorioamnionitis (MC). The purpose of this study is to report the outcomes of asymptomatic neonates ≥35 weeks gestational age (GA) exposed to MC, who were managed without routine antibiotic administration and were clinically monitored while following complete blood cell counts (CBCs). Methods: A retrospective chart review was performed on neonates with GA ≥ 35 weeks with MC during calendar year 2013. IT ratio (immature: total neutrophils) was considered suspicious if ≥0.3. The data were analyzed using independent sample T-tests. Results: Among the 275 neonates with MC, 36 received antibiotics for possible sepsis. Twenty-one were treated with antibiotics for > 48 h for clinical signs of infection; only one infant had a positive blood culture. All 21 became symptomatic prior to initiating antibiotics. Six showed worsening of IT ratio. Thus empiric antibiotic administration was safely avoided in 87% of neonates with MC. 81.5% of the neonates had follow-up appointments within a few days and at two weeks of age within the hospital system. There were no readmissions for suspected sepsis. Conclusions: In our patient population, using CBC indices and clinical observation to predict sepsis in neonates with MC appears safe and avoids the unnecessary use of antibiotics. -

Early-Onset Neonatal Sepsis: a Continuing Problem in Need of Novel Prevention Strategies Barbara J
Early-Onset Neonatal Sepsis: A Continuing Problem in Need of Novel Prevention Strategies Barbara J. Stoll, MD Early-onset neonatal sepsis (EOS) colonized women or targeted IAP for remains a feared cause of severe women with obstetrical risk factors illness and death among infants of all in labor known to increase GBS birthweights and gestational ages, transmission. 5 Revised guidelines with particular impact among preterm in 2002 recommended universal infants. Centers for Disease Control and antenatal screening for GBS at 35 to Prevention investigators have studied 37 weeks’ gestational age to identify the changing epidemiology of invasive colonized women who should receive EOS for several decades. The Active IAP. 6 Guidelines were additionally Bacterial Core surveillance (ABCs) refined in 2010 to provide neonatal network, a collaboration between management recommendations based the Centers for Disease Control and on maternal risk factors and clinical H. Wayne Hightower Distinguished Professor in the Medical Prevention, state health departments, condition of the infant at birth, with Sciences and Dean, McGovern Medical School, University of and universities, was established in an attempt to reduce unnecessary Texas Health Science Center at Houston, Houston, Texas 1995 to address emerging infectious evaluations of well-appearing infants Opinions expressed in these commentaries are diseases of public health importance, without risk factors. 7 Widespread those of the author and not necessarily those of the including infections due to major adherence to national guidelines American Academy of Pediatrics or its Committees. neonatal pathogens. 1, 2 ABCs data resulted in a remarkable decline DOI: 10.1542/peds.2016-3038 are remarkable because of the in early onset GBS disease, but a Accepted for publication Sep 12, 2016 geographic distribution and size of the concomitant increase in exposure Address correspondence to Barbara J. -

Neonatal Sepsis Expanded Tracking Form Instructions
2014 ABCs Neonatal Infection Expanded Tracking Form Instruction Sheet Updated 12/19/2013 This form should be completed for all cases of early- and late-onset group B Streptococcus disease (GBS). Early-onset is defined as GBS disease onset at 0-6 days of age [(culture date-birth date) <7 days]. Late-onset is defined as GBS disease at 7-89 days of age [6 days < (culture date-birth date) <90 days]. This case report form for GBS disease can be completed on infants born at home, but not for stillbirths. Additionally, this form should be filled out for all neonatal sepsis cases, which includes both GBS and non-GBS cases. Neonatal sepsis is defined as invasive bacterial disease onset at 0-2 days of age [(culture date-birth date) <3 days]. Case report forms for neonatal sepsis cases should not be completed on infants born at home or stillbirths. For those sites participating in neonatal sepsis surveillance, please refer to the Neonatal Sepsis protocol for clarification on the inclusion and exclusion criteria. The following is an algorithm of which forms should be filled out for early- & late-onset GBS cases meeting the ABCs case definition: FORMS NNS Surveillance Form Neonatal Infection ABCs Case Report Form SCENARIO Expanded Tracking Form* Early-onset (& Neonatal √ √ √ Sepsis)† Late-onset √ √ *The Neonatal Infection Expanded Tracking Form is the expanded form that combines the Neonatal Sepsis Maternal Case Report Form and the Neonatal group B Streptococcus Disease Prevention Tracking Form. † For CA, CT, GA, and MN, please refer to the Neonatal -

Hypotonia Surestep Product Catalog Page 29 in Step with Pediatric Hypotonia
SPECIAL EDUCATIONAL SERIES DIAGNOSTIC INSIGHTS ANALYZING GAIT CHANGES GROSS MOTOR SKILLS ORTHOTIC MANAGEMENT CLI N I CAL CASE STUDIES Sponsored by an educational grant from: In Step With Pediatric Hypotonia SureStep Product Catalog Page 29 In Step With Pediatric Hypotonia Contents VIEWPOINT FROM THE EDITOR: An Unexpected Path, Mobility and More an Invaluable Perspective At the most basic level, mobility is about get- PAGE 3 ting from point A to point B. But, for many children with hypotonia, it’s about so much 4 more. FEATURES It’s about independence. It’s about con- fidence. It’s about maintaining strength, fit- ness, and healthy bones. It’s about not being Understanding Hypotonia excluded from activities enjoyed by their PAGE 4 typically developing peers. And improved mobility may have even Gait: The Cornerstone more benefits in those children whose hy- potonia is associated with social and behav- of Intervention ioral developmental delays. New research PAGE 8 has identified an association between motor skills and sociobehavioral milestones in chil- 8 The Importance of Gross dren with autism spectrum disorder, who often present with hypotonia (see “The Im- Motor Skills portance of Gross Motor Skills,” page 12). PAGE 12 This suggests that early intervention to improve gross motor skills—including or- thotic devices and physical therapy—may Orthotic Solutions for also help certain children interact more Children with Hypotonia comfortably with others. That won’t come as PAGE 16 a surprise to the clinicians and parents who 12 have personally seen it happen. This special issue is filled with evidence- Orthotic Success Stories: based information and personal success sto- Four Cases in a Series ries illustrating how effective interventions can enhance mobility in children with hy- PAGE 20 potonia. -

Neonatal Pneumonia
Chapter 2 Neonatal Pneumonia Friedrich Reiterer Additional information is available at the end of the chapter http://dx.doi.org/10.5772/54310 1. Introduction Neonatal pneumonia is a serious respiratory infectious disease caused by a variety of microorganisms, mainly bacteria, with the potential of high mortality and morbidity (1,2). Worldwide neonatal pneumonia is estimated to account for up to 10% of childhood mortality, with the highest case fatality rates reported in developing countries (3,4). It´s impact may be increased in the case of early onset, prematurity or an underlying pulmonary condition like RDS, meconium aspiration or CLD/bronchopulmonary dysplasia (BPD), when the pulmonary capacity is already limited. Ureaplasma pneumonia and ventilator- associated pneumonia (VAP) have also been associated with the development of BPD and poor pulmonary outcome (5,6,7). In this chapter we will review different aspects of neonatal pneumonia and will present case reports from our level III neonatal unit in Graz. 2. Epidemiology Reported frequencies of neonatal pneumonia range from 1 to 35 %, the most commonly quoted figures being 1 percent for term infants and 10 percent for preterm infants (8). The incidence varies according to gestational age, intubation status, diagnostic criteria or case definition, the level and standard of neonatal care, race and socioeconomic status. In a retrospective analysis of a cohort of almost 6000 neonates admitted to our NICU pneumonia was diagnosed in all gestational age classes. The incidence of bacterial pneumonia including Ureaplasma urealyticum (Uu) pneumonia was 1,4 % with a median patient gestational age of 35 weeks (range 23-42 weeks) and a mortality of 2,5%. -

The Effects of Maternal Chorioamnionitis on the Neonate
Neonatal Nursing Education Brief: The Effects of Maternal Chorioamnionitis on the Neonate https://www.seattlechildrens.org/healthcare- professionals/education/continuing-medical-nursing-education/neonatal- nursing-education-briefs/ Maternal chorioamnionitis is a common condition that can have negative effects on the neonate. The use of broad spectrum antibiotics in labor can reduce the risks, but infants exposed to chorioamnionitis continue to require treatment. The neonatal sepsis risk calculator can guide treatment. NICU, chorioamnionitis, early onset neonatal sepsis, sepsis risk calculator The Effects of Maternal Chorioamnionitis on the Neonate Purpose and Goal: CNEP # 2090 • Understand the effects of chorioamnionitis on the neonate. • Learn about a new approach for treating infants at risk. None of the planners, faculty or content specialists has any conflict of interest or will be presenting any off-label product use. This presentation has no commercial support or sponsorship, nor is it co-sponsored. Requirements for successful completion: • Successfully complete the post-test • Complete the evaluation form Date • December 2018 – December 2020 Learning Objectives • Describe the pathogenesis of maternal chorioamnionitis. • Describe the outcomes for neonates exposed to chorioamnionitis. • Identify 2 approaches for the treatment of early onset sepsis. Introduction • Chorioamnionitis is a common complication • It affects up to 10% of all pregnancies • It is an infection of the amniotic fluid and placenta • It is characterized by inflammation -

Neonatal Sepsis Management Guidelines (PDF)
Clinical Policy: Neonatal Sepsis Management Reference Number: CP.MP.85 Date of Last Revision: 07/21 Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Through the increased incidence of intra-partum antibiotics, early-onset neonatal sepsis is occurring less frequently. However, it continues to be a common cause of neonatal morbidity and mortality. The CDC (Centers for Disease Control and Prevention) defines early onset sepsis as a blood or cerebrospinal fluid culture-proven infection occurring within the first seven days of life. Group B Streptococcus (GBS) remains the leading cause of neonatal sepsis. More than half of GBS cases occur in infants of mothers with negative GBS cultures, emphasizing the need to remain vigilant for signs of sepsis in all newborns. These infants require comprehensive assessment and treatment, as well as discharge planning, in order to ensure timely treatment in an effort to reduce morbidity and mortality.2 Policy/Criteria It is the policy of Health Plans affiliated with Centene Corporation that the management of neonatal sepsis is medically necessary at the indicated level of care for the following circumstances: I. Episode Day 1 A. Well-appearing infants who are on 48 hours of antibiotics pending blood culture results are appropriate for level II (rev code 172) nursery. B. Symptomatic infants are appropriate for level III (rev code 173) nursery with all the following: 1. Hypotonia, lethargy, or poor oral feeding; 2. Temp ≥ 100.4◦F or ≤ 96.8◦F (≥ 38.0◦ or ≤ 36.0◦C); 3. On 48 hours of antibiotics pending blood culture results or treatment of positive blood cultures. -
IUGR: Intrauterine Growth Restriction
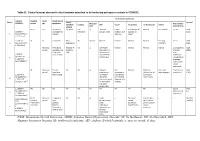
Table S1. Clinical features observed in the 6 patients described so far harboring pathogenic variants in FOXRED1. Evolutionary symptoms Variants Prenatal Onset Onset clinical Patient Lactic/ Survival FOXRED1 period age symptoms Muscular Psychomotor Metabolic Epilepsy MRI Visual Respiratory Cardiovascular Others tone development acidosis IUGR 2m Hypotonia Yes (+++) Yes ↓ Normal Latent Bronchospasm Normal AEP normal IQ: 48 Alive c.920G>A Development refractary (2m,4y,7y3m) strabismus of episodes in (15y) 1 (p.Gly307Glu) / al delay right eye infant c.733+1G>A c.920G>A NI 4y Clumsiness With No Normal Normal Normal Normal Normal Learning IQ: 99 Alive 2 (p.Gly307Glu) / exercise (+) disorders (19y) c.733+1G>A - Neonatal Premature; No (only ↑ Yes ↓ Decreased Normal Normal Normal Normal Gradually loss Alive period Hypoglycemia lactate in attenuation in of motor (22y)) Congenital LCR) the putamen skills; c.694C>T lactic acidosis and cerebellar wheelchair; (p.Gln232X) / 3 atrophy (6y) no expressive c.1289A>G language; 18 (p.Asn430Ser) understands simple commands NI Neonatal Truncal Yes Yes ↓ Delayed Eye Normal Mild non- Persistent Psychomotor Alive period hypotonia myelination movements obstructive left hepatomegaly retardation (10y) c.1054 C>T Poor feeding ventricular have always ventricular (p.Arg352Trp) / dilatation; been roving hypertrophy 4 c.1054 C>T abnormal signal bilateral optic (p.Arg352Trp) 19 in the thalami atrophy and basal ganglia (8m) c.1308G>A ND ND ND ND Yes NA NA NA NA NA NA Severe Alive (p.Val421Met) / psychomotor (¿) 5 c.1308G>A retardation (p.Val421Met)20 IUGR; Neonatal Congenital Yes Yes ↓ Large -- Persistent Dilated right - - Death (3 c.612_615dupA Cerebral period lactic periventricular severe ventricle and months) CTG intraventric acidosis. -

Cerebral Hypotonia by Mihee Bay MD (Dr
Cerebral hypotonia By Mihee Bay MD (Dr. Bay of Kennedy Krieger Institute and Johns Hopkins School of Medicine has no relevant financial relationships to disclose.) Originally released July 12, 2006; last updated February 1, 2016; expires February 1, 2019 Introduction This article includes discussion of cerebral hypotonia, central hypotonia, essential hypotonia, benign congenital hypotonia, and floppy infant. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations. Overview Hypotonia is a clinical manifestation of numerous diseases affecting the central and/or peripheral motor nervous system. The key to accurate diagnosis involves integral steps of evaluation that include a detailed history, examination, and diagnostic tests. “Cerebral” (or central) hypotonia implies pathogenesis from abnormalities from the central nervous system, and related causal disorders include cerebral dysgenesis and genetic or metabolic disorders. Patients with central hypotonia generally have hypotonia without associated weakness, in contrast to the peripheral (lower motor neuron) causes, which typically produce both hypotonia and muscle weakness. Hypotonia is a clinical manifestation of over 500 genetic disorders; thus, a logical, stepwise approach to diagnosis is essential. With recent advances in the field of genetic testing, diagnostic yield will undoubtedly improve. There is no cure, but treatment includes supportive therapies, such as physical and occupational therapy, and diagnosis-specific management. Key points • Hypotonia is reduced tension or resistance of passive range of motion. • The first step in the evaluation of a child with hypotonia is localization to the central (“cerebral”) or peripheral nervous system, or both. • Central hypotonia is more likely to be noted axially with normal strength and hyperactive to normal deep tendon reflexes. -

Antibiotic Use for Sepsis in Neonates and Children: 2016 Evidence Update
Antibiotic Use for Sepsis in Neonates and Children: 2016 Evidence Update Aline Fuchsa, Julia Bielickia,b, Shrey Mathurb, Mike Sharlandb, Johannes N. Van Den Ankera,c a Paediatric Pharmacology and Pharmacometrics, University Children's Hospital Basel, Basel, Switzerland b Paediatric Infectious Disease Research Group, Institute for Infection and Immunity, St George's University of London, London, United Kingdom c Division of Clinical Pharmacology, Children’s National Health System, Washington, DC, USA WHO-Reviews 1 TABLE OF CONTENTS 1. INTRODUCTION ............................................................................................................................... 3 1.1. Aims ......................................................................................................................................... 3 1.2. Background ............................................................................................................................. 3 1.2.1. Definition and diagnosis ................................................................................................. 3 Neonatal Sepsis ............................................................................................................................... 3 Paediatric Sepsis ............................................................................................................................. 4 Community versus hospital acquired sepsis .................................................................................. 5 1.2.2. Microbiology .................................................................................................................. -

Mean Platelet Volume in Asymptomatic Chorioamnionitis-Exposed Infants
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2021;10(1):e100132 doi: 10.7363/100132 Received: 2019 Aug 22; revised: 2020 Jan 26; accepted: 2020 Feb 02; published online: 2020 Dec 28 Original article Mean platelet volume in asymptomatic chorioamnionitis- exposed infants. A retrospective case-control study Atef Alshafei, Moustafa Hassan, Yaser El saba, Anwar Khan, Mahmoud Ahmed Neonatology Section, Pediatric Department, Dubai Hospital, Dubai, UAE Abstract Introduction: Maternal chorioamnionitis (CA) is a serious condition causing several neonatal morbidities and long-term neurodevelopmental sequelae in exposed infants. Current guidelines still recommend admission, laboratory evaluation, and antibiotic administration to all CA-exposed infants. The incidence of early-onset neonatal sepsis (EOS) is currently low, owing to the routine intrapartum antibiotic administration to mothers identified to be at risk of developing CA. New diagnostic tools for early diagnosis of sepsis in apparently healthy infants exposed to maternal CA are needed. Previous studies showed that mean platelet volume (MPV) is evolving as a potential inflammatory marker of neonatal sepsis. We aimed to study whether MPV can be used as an adjuvant diagnostic tool for EOS in asymptomatic CA-exposed infants. Objective: To evaluate the role of MPV as an adjuvant biomarker of EOS in cases of asymptomatic CA-exposed infants. Design: Retrospective case-control study. Setting: A tertiary care Neonatal Intensive Care Unit (NICU). Patients: Asymptomatic CA-exposed infants 37-40 weeks of gestation admitted between May 2016 and April 2019 to the NICU of Dubai Hospital, UAE. Results: A total of 1,300 infants were admitted to NICU during the study period. -

Best Practices to Addressing Early Onset Neonatal Sepsis Rakhi Gupta Basuray, MD, FAAP Ansley Splinter, MD, Macm, FAAP Objective
Best Practices to Addressing Early Onset Neonatal Sepsis Rakhi Gupta Basuray, MD, FAAP Ansley Splinter, MD, MAcM, FAAP Objective Apply clinical guidelines for the management of neonates with suspected or proven early onset sepsis Early Onset Sepsis - Defined • Culture-proven, invasive infection, within the first 72 HOL • Mostly due to ascension from normal maternal GI / GU flora • GBS ~40%, E coli ~25% • Current incidence of GBS EOS 0.25/1000 (≥34 weeks GA) • Incidence of all causes of EOS 0.5/1000 Success of Intrapartum Antibiotic Prophylaxis • Since 1995 • 85% decline in GBS EOS, as of 2010 • Used in ~30% of births • No impact on late onset neonatal sepsis 2010 CDC Guidelines • Risk based • Yes/No dichotomous classifications • High weight given to chorioamnionitis • If YES = Antibiotic Treatment MMWR (2010) Vol.59/No.RR-10 2012 AAP Guidelines Evaluation of asymptomatic infants 37 weeks or greater with risk factors for sepsis Polin and the COFN Pediatrics 2012;129:1006-1015 How Well Do Yes/No Risk Factors Perform At Finding Infected Infants? Puopolo KM, et al. Pediatrics. 2011 Nov;128(5):e1155-63 2010 CDC Gaps • Based on EOS incidences 5-10x higher than current • Management guided by risk factors • Chorioamnionitis diagnosis inconsistent • Clinical signs of illness not defined • CBC, CRP interpretations non-standard **200 neonates treated with antibiotics for each episode of EOS** Sepsis Risk Calculator • Puopolo & Escobar • Risk Prediction Tool (2011) • Refined with infant exam findings (2014) • Prospectively validated (2017) https://neonatalsepsiscalculator.kaiserpermanente.org