Review and Hypothesis: Syndromes with Severe Intrauterine Growth
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical Utility Gene Card For: 3-M Syndrome – Update 2013
European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: 3-M syndrome – Update 2013 Muriel Holder-Espinasse*,1, Melita Irving1 and Vale´rie Cormier-Daire2 European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156; published online 31 July 2013 Update to: European Journal of Human Genetics (2011) 19, doi:10.1038/ejhg.2011.32; published online 2 March 2011 1. DISEASE CHARACTERISTICS nonsense and missense mutations c.4333C4T (p.Arg1445*) and 1.1 Name of the disease (synonyms) c.4391A4C (p.His1464Pro), respectively, render CUL7 deficient 3-M syndrome (gloomy face syndrome, dolichospondylic dysplasia). in recruiting ROC1, leading to impaired ubiquitination. OBSL1: microsatellites analysis of the locus (2q35-36.1) in con- 1.2 OMIM# of the disease sanguineous families. OBSL1: microsatellites analysis of the locus 273750. (2q35-36.1) in consanguineous families. Mutations induce non- sense mediated decay. Knockdown of OBSL1 in HEK293 cells 1.3 Name of the analysed genes or DNA/chromosome segments shows the role of this gene in the maintenance of normal levels of CUL7, OBSL1 and CCDC8.1–5 CUL7. Abnormal IGFBP2 andIGFBP5 mRNA levels in two patients with OBSL1 mutations, suggesting that OBSL1 modulates the 1.4 OMIM# of the gene(s) expression of IGFBP proteins. CCDC8: microsatellites analysis 609577 (CUL7), 610991 (OBSL1) and 614145 (CCDC8). at the locus (19q13.2-q13.32). CCDC8, 1-BP DUP, 612G and CCDC8, 1-BP. -

Stillbirths Preceded by Reduced Fetal Movements Are More Frequently Associated with Placental Insufficiency: a Retrospective Cohort Study
J. Perinat. Med. 2021; aop Madeleine ter Kuile, Jan Jaap H.M. Erwich and Alexander E.P. Heazell* Stillbirths preceded by reduced fetal movements are more frequently associated with placental insufficiency: a retrospective cohort study https://doi.org/10.1515/jpm-2021-0103 RFM were less frequently reported in twin pregnancies Received March 3, 2021; accepted June 25, 2021; ending in stillbirth and in intrapartum stillbirths. published online July 15, 2021 Conclusions: The association between RFM and placental insufficiency was confirmed in cases of stillbirth. This Abstract provides further evidence that RFM is a symptom of placental insufficiency. Therefore, investigation after RFM Objectives: Maternal report of reduced fetal movements should aim to identify placental dysfunction. (RFM) is a means of identifying fetal compromise in preg- nancy. In live births RFM is associated with altered Keywords: absent fetal movement; decreased fetal move- placental structure and function. Here, we explored asso- ment; perinatal mortality; placenta. ciations between RFM, pregnancy characteristics, and the presence of placental abnormalities and fetal growth re- striction (FGR) in cases of stillbirth. Introduction Methods: A retrospective cohort study was carried out in a single UK tertiary maternity unit. Cases were divided into Stillbirth is an extensive problem that receives little atten- three groups: 109 women reporting RFM, 33 women with tion from worldwide initiatives [1]. Although only 2% of the absent fetal movements (AFM) and 159 who did not report 2.8 million stillbirths each year occur in high-income RFM before the diagnosis of stillbirth. Univariate and countries (HICs), this still accounts for significant number multivariate logistic regression was used to determine as- of deaths [2]. -

Patient Advocacy Organizations, Industry Funding, and Conflicts of Interest
Supplementary Online Content Rose SL, Highland J, Karafa MT, Joffe S. Patient advocacy organizations, industry funding, and conflicts of interest. JAMA Intern Med. Published online January 17, 2017. doi:10.1001/jamainternmed.2016.8443 eTable 1. Search Terms Used to Identify Organizations eTable 2. Inclusion and Exclusion Criteria eTable 3. Sensitivity Analysis Comparing Survey Responses of Executive and Non-Executive Respondents of Patient Advocacy Organizations (PAOs) eFigure. Survey This supplementary material has been provided by the authors to give readers additional information about their work. © 2017 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable 1. Search Terms Used to Identify Organizations Aagenaes Syndrome Anoxia Carcinoid Aarskog Syndrome Antiphospholipid Syndrome Carcinoma Aase-Smith Syndrome II Antley-Bixler Syndrome Cardiac Abdominal Cystic Phenotype Cardiogenic Lymphangioma Anxiety Cardiogenic Shock Abdominal Obesity Metabolic Aphasia Cardiomyopathy Syndrome Apraxia Cardiovascular Achondroplasia Arrhythmia Carotid Artery Disease Achromatopsia Arteriosclerosis Celiac Acid Lipase Disease Arthritis Central Cord Syndrome Acoustic Neuroma Asperger Syndrome Cervical Cancer Acquired Hyperostosis Aspergers Chondrodysplasia Punctata Syndrome Asthma Acrocephalosyndactylia Chordoma Astrocytoma Addison Disease Churg-Strauss Syndrome Ataxia ADHD Colon Cancer Atherosclerosis Adie's syndrome Colorectal Cancer Atrial Adrenal Hyperplasia Conduct Disorder Atrial Fibulation -

Trim37-Deficient Mice Recapitulate Several Features of the Multi-Organ
© 2016. Published by The Company of Biologists Ltd | Biology Open (2016) 5, 584-595 doi:10.1242/bio.016246 RESEARCH ARTICLE Trim37-deficient mice recapitulate several features of the multi- organ disorder Mulibrey nanism Kaisa M. Kettunen1,2,3,4, Riitta Karikoski5, Riikka H. Hämäläinen6, Teija T. Toivonen1, Vasily D. Antonenkov7, Natalia Kulesskaya3, Vootele Voikar3, Maarit Hölttä-Vuori8,9, Elina Ikonen8,9, Kirsi Sainio10, Anu Jalanko11, Susann Karlberg12, Niklas Karlberg12, Marita Lipsanen-Nyman12, Jorma Toppari13, Matti Jauhiainen11, J. Kalervo Hiltunen7, Hannu Jalanko14 and Anna-Elina Lehesjoki1,2,3,* ABSTRACT of the human MUL disease and thus provide a good model to study Mulibrey nanism (MUL) is a rare autosomal recessive multi-organ disease pathogenesis related to TRIM37 deficiency, including disorder characterized by severe prenatal-onset growth failure, infertility, non-alcoholic fatty liver disease, cardiomyopathy and infertility, cardiopathy, risk for tumors, fatty liver, and type 2 tumorigenesis. diabetes. MUL is caused by loss-of-function mutations in TRIM37, KEY WORDS: Mulibrey nanism, Infertility, Fatty liver, which encodes an E3 ubiquitin ligase belonging to the tripartite motif Cardiomyopathy, Tumorigenesis, Growth failure (TRIM) protein family and having both peroxisomal and nuclear localization. We describe a congenic Trim37 knock-out mouse INTRODUCTION (Trim37−/−) model for MUL. Trim37−/− mice were viable and had Mulibrey nanism (MUL) is a rare autosomal recessive multi-organ normal weight development until approximately -

Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H
Hypotonia and Lethargy in a Two-Day-Old Male Infant Adrienne H. Long, MD, PhD,a,b Jennifer G. Fiore, MD,a,b Riaz Gillani, MD,a,b Laurie M. Douglass, MD,c Alan M. Fujii, MD,d Jodi D. Hoffman, MDe A 2-day old term male infant was found to be hypotonic and minimally abstract reactive during routine nursing care in the newborn nursery. At 40 hours of life, he was hypoglycemic and had intermittent desaturations to 70%. His mother had an unremarkable pregnancy and spontaneous vaginal delivery. The mother’s prenatal serology results were negative for infectious risk factors. Apgar scores were 9 at 1 and 5 minutes of life. On day 1 of life, he fed, stooled, and voided well. Our expert panel discusses the differential diagnosis of hypotonia in a neonate, offers diagnostic and management recommendations, and discusses the final diagnosis. DRS LONG, FIORE, AND GILLANI, birth weight was 3.4 kg (56th PEDIATRIC RESIDENTS percentile), length was 52 cm (87th aDepartment of Medicine, Boston Children’s Hospital, d e percentile), and head circumference Boston, Massachusetts; and Neonatology Section, Medical A 2-day old male infant born at Genetics Section, cDivision of Child Neurology, and 38 weeks and 4 days was found to be was 33 cm (12th percentile). His bDepartment of Pediatrics, Boston Medical Center, Boston, limp and minimally reactive during physical examination at birth was Massachusetts routine care in the newborn nursery. normal for gestational age, with Drs Long, Fiore, and Gillani conceptualized, drafted, Just 5 hours before, he had an appropriate neurologic, cardiac, and and edited the manuscript; Drs Douglass, Fujii, and appropriate neurologic status when respiratory components. -

Repercussions of Inborn Errors of Immunity on Growth☆ Jornal De Pediatria, Vol
Jornal de Pediatria ISSN: 0021-7557 ISSN: 1678-4782 Sociedade Brasileira de Pediatria Goudouris, Ekaterini Simões; Segundo, Gesmar Rodrigues Silva; Poli, Cecilia Repercussions of inborn errors of immunity on growth☆ Jornal de Pediatria, vol. 95, no. 1, Suppl., 2019, pp. S49-S58 Sociedade Brasileira de Pediatria DOI: https://doi.org/10.1016/j.jped.2018.11.006 Available in: https://www.redalyc.org/articulo.oa?id=399759353007 How to cite Complete issue Scientific Information System Redalyc More information about this article Network of Scientific Journals from Latin America and the Caribbean, Spain and Journal's webpage in redalyc.org Portugal Project academic non-profit, developed under the open access initiative J Pediatr (Rio J). 2019;95(S1):S49---S58 www.jped.com.br REVIEW ARTICLE ଝ Repercussions of inborn errors of immunity on growth a,b,∗ c,d e Ekaterini Simões Goudouris , Gesmar Rodrigues Silva Segundo , Cecilia Poli a Universidade Federal do Rio de Janeiro (UFRJ), Faculdade de Medicina, Departamento de Pediatria, Rio de Janeiro, RJ, Brazil b Universidade Federal do Rio de Janeiro (UFRJ), Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Curso de Especializac¸ão em Alergia e Imunologia Clínica, Rio de Janeiro, RJ, Brazil c Universidade Federal de Uberlândia (UFU), Faculdade de Medicina, Departamento de Pediatria, Uberlândia, MG, Brazil d Universidade Federal de Uberlândia (UFU), Hospital das Clínicas, Programa de Residência Médica em Alergia e Imunologia Pediátrica, Uberlândia, MG, Brazil e Universidad del Desarrollo, -

2021 Western Medical Research Conference
Abstracts J Investig Med: first published as 10.1136/jim-2021-WRMC on 21 December 2020. Downloaded from Genetics I Purpose of Study Genomic sequencing has identified a growing number of genes associated with developmental brain disorders Concurrent session and revealed the overlapping genetic architecture of autism spectrum disorder (ASD) and intellectual disability (ID). Chil- 8:10 AM dren with ASD are often identified first by psychologists or neurologists and the extent of genetic testing or genetics refer- Friday, January 29, 2021 ral is variable. Applying clinical whole genome sequencing (cWGS) early in the diagnostic process has the potential for timely molecular diagnosis and to circumvent the diagnostic 1 PROSPECTIVE STUDY OF EPILEPSY IN NGLY1 odyssey. Here we report a pilot study of cWGS in a clinical DEFICIENCY cohort of young children with ASD. RJ Levy*, CH Frater, WB Galentine, MR Ruzhnikov. Stanford University School of Medicine, Methods Used Children with ASD and cognitive delays/ID Stanford, CA were referred by neurologists or psychologists at a regional healthcare organization. Medical records were used to classify 10.1136/jim-2021-WRMC.1 probands as 1) ASD/ID or 2) complex ASD (defined as 1 or more major malformations, abnormal head circumference, or Purpose of Study To refine the electroclinical phenotype of dysmorphic features). cWGS was performed using either epilepsy in NGLY1 deficiency via prospective clinical and elec- parent-child trio (n=16) or parent-child-affected sibling (multi- troencephalogram (EEG) findings in an international cohort. plex families; n=3). Variants were classified according to Methods Used We performed prospective phenotyping of 28 ACMG guidelines. -

Hypotonia Surestep Product Catalog Page 29 in Step with Pediatric Hypotonia
SPECIAL EDUCATIONAL SERIES DIAGNOSTIC INSIGHTS ANALYZING GAIT CHANGES GROSS MOTOR SKILLS ORTHOTIC MANAGEMENT CLI N I CAL CASE STUDIES Sponsored by an educational grant from: In Step With Pediatric Hypotonia SureStep Product Catalog Page 29 In Step With Pediatric Hypotonia Contents VIEWPOINT FROM THE EDITOR: An Unexpected Path, Mobility and More an Invaluable Perspective At the most basic level, mobility is about get- PAGE 3 ting from point A to point B. But, for many children with hypotonia, it’s about so much 4 more. FEATURES It’s about independence. It’s about con- fidence. It’s about maintaining strength, fit- ness, and healthy bones. It’s about not being Understanding Hypotonia excluded from activities enjoyed by their PAGE 4 typically developing peers. And improved mobility may have even Gait: The Cornerstone more benefits in those children whose hy- potonia is associated with social and behav- of Intervention ioral developmental delays. New research PAGE 8 has identified an association between motor skills and sociobehavioral milestones in chil- 8 The Importance of Gross dren with autism spectrum disorder, who often present with hypotonia (see “The Im- Motor Skills portance of Gross Motor Skills,” page 12). PAGE 12 This suggests that early intervention to improve gross motor skills—including or- thotic devices and physical therapy—may Orthotic Solutions for also help certain children interact more Children with Hypotonia comfortably with others. That won’t come as PAGE 16 a surprise to the clinicians and parents who 12 have personally seen it happen. This special issue is filled with evidence- Orthotic Success Stories: based information and personal success sto- Four Cases in a Series ries illustrating how effective interventions can enhance mobility in children with hy- PAGE 20 potonia. -

Smells Like Teen Spirit Appears in Rock & Pop 2018
ACCESS ALL AREAS... SMELLS LIKE TEEN SPIRIT APPEARS IN ROCK & POP 2018 Released: 1991 Album: Nevermind Label: DGC Records ABOUT THE SONG Nirvana’s Kurt Cobain was attempting to write the ‘ultimate pop song’ when he came up with the guitar riff that would become ‘Smells Like Teen Spirit’. He WITH THE LIGHTS OUT, wanted to write a song in the style of The Pixies, telling Rolling Stone in 1994: ‘I was basically trying to rip off The Pixies. I have to admit it.’ The title came IT’S LESS DANGEROUS after Kathleen Hanna, lead singer of Bikini Kill, spray- painted ‘Kurt smells like Teen Spirit’ on his bedroom “ wall. Teen Spirit was actually a brand of deodorant. HERE WE ARE NOW, The first single from Nirvana’s second album Nevermind, ‘Smells Like Teen Spirit’ was a surprise hit. The label had anticipated that ‘Come As You Are’, the follow-up single, would be the song to cross over to a mainstream audience. ‘Smells Like Teen Spirit’ ENTERTAIN US was first played on college radio before rock stations and MTV picked it up. It is widely praised as one of I FEEL STUPID AND CONTAGIOUS the greatest songs in the history of rock music. RECORDING AND PRODUCTION Cobain began writing ‘Smells Like Teen Spirit’ a few weeks before Nirvana were due in the studio to record Nevermind. After presenting the main riff and melody of the chorus to the rest of the band, they jammed around the riff for an hour and a half. Bassist Krist Novoselic slowed the verse down and drummer Dave Grohl created a drum beat and as a result, ‘Smells Like Teen Spirit’ is the only song on Nevermind to give songwriting credits to all three band members. -
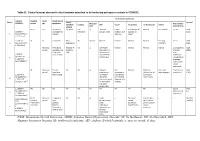
IUGR: Intrauterine Growth Restriction
Table S1. Clinical features observed in the 6 patients described so far harboring pathogenic variants in FOXRED1. Evolutionary symptoms Variants Prenatal Onset Onset clinical Patient Lactic/ Survival FOXRED1 period age symptoms Muscular Psychomotor Metabolic Epilepsy MRI Visual Respiratory Cardiovascular Others tone development acidosis IUGR 2m Hypotonia Yes (+++) Yes ↓ Normal Latent Bronchospasm Normal AEP normal IQ: 48 Alive c.920G>A Development refractary (2m,4y,7y3m) strabismus of episodes in (15y) 1 (p.Gly307Glu) / al delay right eye infant c.733+1G>A c.920G>A NI 4y Clumsiness With No Normal Normal Normal Normal Normal Learning IQ: 99 Alive 2 (p.Gly307Glu) / exercise (+) disorders (19y) c.733+1G>A - Neonatal Premature; No (only ↑ Yes ↓ Decreased Normal Normal Normal Normal Gradually loss Alive period Hypoglycemia lactate in attenuation in of motor (22y)) Congenital LCR) the putamen skills; c.694C>T lactic acidosis and cerebellar wheelchair; (p.Gln232X) / 3 atrophy (6y) no expressive c.1289A>G language; 18 (p.Asn430Ser) understands simple commands NI Neonatal Truncal Yes Yes ↓ Delayed Eye Normal Mild non- Persistent Psychomotor Alive period hypotonia myelination movements obstructive left hepatomegaly retardation (10y) c.1054 C>T Poor feeding ventricular have always ventricular (p.Arg352Trp) / dilatation; been roving hypertrophy 4 c.1054 C>T abnormal signal bilateral optic (p.Arg352Trp) 19 in the thalami atrophy and basal ganglia (8m) c.1308G>A ND ND ND ND Yes NA NA NA NA NA NA Severe Alive (p.Val421Met) / psychomotor (¿) 5 c.1308G>A retardation (p.Val421Met)20 IUGR; Neonatal Congenital Yes Yes ↓ Large -- Persistent Dilated right - - Death (3 c.612_615dupA Cerebral period lactic periventricular severe ventricle and months) CTG intraventric acidosis. -

Cerebral Hypotonia by Mihee Bay MD (Dr
Cerebral hypotonia By Mihee Bay MD (Dr. Bay of Kennedy Krieger Institute and Johns Hopkins School of Medicine has no relevant financial relationships to disclose.) Originally released July 12, 2006; last updated February 1, 2016; expires February 1, 2019 Introduction This article includes discussion of cerebral hypotonia, central hypotonia, essential hypotonia, benign congenital hypotonia, and floppy infant. The foregoing terms may include synonyms, similar disorders, variations in usage, and abbreviations. Overview Hypotonia is a clinical manifestation of numerous diseases affecting the central and/or peripheral motor nervous system. The key to accurate diagnosis involves integral steps of evaluation that include a detailed history, examination, and diagnostic tests. “Cerebral” (or central) hypotonia implies pathogenesis from abnormalities from the central nervous system, and related causal disorders include cerebral dysgenesis and genetic or metabolic disorders. Patients with central hypotonia generally have hypotonia without associated weakness, in contrast to the peripheral (lower motor neuron) causes, which typically produce both hypotonia and muscle weakness. Hypotonia is a clinical manifestation of over 500 genetic disorders; thus, a logical, stepwise approach to diagnosis is essential. With recent advances in the field of genetic testing, diagnostic yield will undoubtedly improve. There is no cure, but treatment includes supportive therapies, such as physical and occupational therapy, and diagnosis-specific management. Key points • Hypotonia is reduced tension or resistance of passive range of motion. • The first step in the evaluation of a child with hypotonia is localization to the central (“cerebral”) or peripheral nervous system, or both. • Central hypotonia is more likely to be noted axially with normal strength and hyperactive to normal deep tendon reflexes. -

Mean Platelet Volume in Asymptomatic Chorioamnionitis-Exposed Infants
www.jpnim.com Open Access eISSN: 2281-0692 Journal of Pediatric and Neonatal Individualized Medicine 2021;10(1):e100132 doi: 10.7363/100132 Received: 2019 Aug 22; revised: 2020 Jan 26; accepted: 2020 Feb 02; published online: 2020 Dec 28 Original article Mean platelet volume in asymptomatic chorioamnionitis- exposed infants. A retrospective case-control study Atef Alshafei, Moustafa Hassan, Yaser El saba, Anwar Khan, Mahmoud Ahmed Neonatology Section, Pediatric Department, Dubai Hospital, Dubai, UAE Abstract Introduction: Maternal chorioamnionitis (CA) is a serious condition causing several neonatal morbidities and long-term neurodevelopmental sequelae in exposed infants. Current guidelines still recommend admission, laboratory evaluation, and antibiotic administration to all CA-exposed infants. The incidence of early-onset neonatal sepsis (EOS) is currently low, owing to the routine intrapartum antibiotic administration to mothers identified to be at risk of developing CA. New diagnostic tools for early diagnosis of sepsis in apparently healthy infants exposed to maternal CA are needed. Previous studies showed that mean platelet volume (MPV) is evolving as a potential inflammatory marker of neonatal sepsis. We aimed to study whether MPV can be used as an adjuvant diagnostic tool for EOS in asymptomatic CA-exposed infants. Objective: To evaluate the role of MPV as an adjuvant biomarker of EOS in cases of asymptomatic CA-exposed infants. Design: Retrospective case-control study. Setting: A tertiary care Neonatal Intensive Care Unit (NICU). Patients: Asymptomatic CA-exposed infants 37-40 weeks of gestation admitted between May 2016 and April 2019 to the NICU of Dubai Hospital, UAE. Results: A total of 1,300 infants were admitted to NICU during the study period.