Lab 02: How to Handle Phylogenetic
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 A) Login to the System B) Use the Appropriate Command to Determine Your Login Shell C) Use the /Etc/Passwd File to Verify the Result of Step B
CSE ([email protected] II-Sem) EXP-3 1 a) Login to the system b) Use the appropriate command to determine your login shell c) Use the /etc/passwd file to verify the result of step b. d) Use the ‘who’ command and redirect the result to a file called myfile1. Use the more command to see the contents of myfile1. e) Use the date and who commands in sequence (in one line) such that the output of date will display on the screen and the output of who will be redirected to a file called myfile2. Use the more command to check the contents of myfile2. 2 a) Write a “sed” command that deletes the first character in each line in a file. b) Write a “sed” command that deletes the character before the last character in each line in a file. c) Write a “sed” command that swaps the first and second words in each line in a file. a. Log into the system When we return on the system one screen will appear. In this we have to type 100.0.0.9 then we enter into editor. It asks our details such as Login : krishnasai password: Then we get log into the commands. bphanikrishna.wordpress.com FOSS-LAB Page 1 of 10 CSE ([email protected] II-Sem) EXP-3 b. use the appropriate command to determine your login shell Syntax: $ echo $SHELL Output: $ echo $SHELL /bin/bash Description:- What is "the shell"? Shell is a program that takes your commands from the keyboard and gives them to the operating system to perform. -
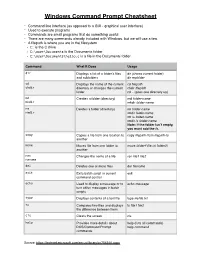
Windows Command Prompt Cheatsheet
Windows Command Prompt Cheatsheet - Command line interface (as opposed to a GUI - graphical user interface) - Used to execute programs - Commands are small programs that do something useful - There are many commands already included with Windows, but we will use a few. - A filepath is where you are in the filesystem • C: is the C drive • C:\user\Documents is the Documents folder • C:\user\Documents\hello.c is a file in the Documents folder Command What it Does Usage dir Displays a list of a folder’s files dir (shows current folder) and subfolders dir myfolder cd Displays the name of the current cd filepath chdir directory or changes the current chdir filepath folder. cd .. (goes one directory up) md Creates a folder (directory) md folder-name mkdir mkdir folder-name rm Deletes a folder (directory) rm folder-name rmdir rmdir folder-name rm /s folder-name rmdir /s folder-name Note: if the folder isn’t empty, you must add the /s. copy Copies a file from one location to copy filepath-from filepath-to another move Moves file from one folder to move folder1\file.txt folder2\ another ren Changes the name of a file ren file1 file2 rename del Deletes one or more files del filename exit Exits batch script or current exit command control echo Used to display a message or to echo message turn off/on messages in batch scripts type Displays contents of a text file type myfile.txt fc Compares two files and displays fc file1 file2 the difference between them cls Clears the screen cls help Provides more details about help (lists all commands) DOS/Command Prompt help command commands Source: https://technet.microsoft.com/en-us/library/cc754340.aspx. -

How to Find out the IP Address of an Omron
Communications Middleware/Network Browser How to find an Omron Controller’s IP address Valin Corporation | www.valin.com Overview • Many Omron PLC’s have Ethernet ports or Ethernet port options • The IP address for a PLC is usually changed by the programmer • Most customers do not mark the controller with IP address (label etc.) • Very difficult to communicate to the PLC over Ethernet if the IP address is unknown. Valin Corporation | www.valin.com Simple Ethernet Network Basics IP address is up to 12 digits (4 octets) Ex:192.168.1.1 For MOST PLC programming applications, the first 3 octets are the network address and the last is the node address. In above example 192.168.1 is network address, 1 is node address. For devices to communicate on a simple network: • Every device IP Network address must be the same. • Every device node number must be different. Device Laptop EX: Omron PLC 192.168.1.1 192.168.1.1 Device Laptop EX: Omron PLC 127.27.250.5 192.168.1.1 Device Laptop EX: Omron PLC 192.168.1.3 192.168.1.1 Valin Corporation | www.valin.com Omron Default IP Address • Most Omron Ethernet devices use one of the following IP addresses by default. Omron PLC 192.168.250.1 OR 192.168.1.1 Valin Corporation | www.valin.com PING Command • PING is a way to check if the device is connected (both virtually and physically) to the network. • Windows Command Prompt command. • PC must use the same network number as device (See previous) • Example: “ping 172.21.90.5” will test to see if a device with that IP address is connected to the PC. -

Disk Clone Industrial
Disk Clone Industrial USER MANUAL Ver. 1.0.0 Updated: 9 June 2020 | Contents | ii Contents Legal Statement............................................................................... 4 Introduction......................................................................................4 Cloning Data.................................................................................................................................... 4 Erasing Confidential Data..................................................................................................................5 Disk Clone Overview.......................................................................6 System Requirements....................................................................................................................... 7 Software Licensing........................................................................................................................... 7 Software Updates............................................................................................................................. 8 Getting Started.................................................................................9 Disk Clone Installation and Distribution.......................................................................................... 12 Launching and initial Configuration..................................................................................................12 Navigating Disk Clone.....................................................................................................................14 -

Don't Trust Traceroute (Completely)
Don’t Trust Traceroute (Completely) Pietro Marchetta, Valerio Persico, Ethan Katz-Bassett Antonio Pescapé University of Southern California, CA, USA University of Napoli Federico II, Italy [email protected] {pietro.marchetta,valerio.persico,pescape}@unina.it ABSTRACT In this work, we propose a methodology based on the alias resolu- tion process to demonstrate that the IP level view of the route pro- vided by traceroute may be a poor representation of the real router- level route followed by the traffic. More precisely, we show how the traceroute output can lead one to (i) inaccurately reconstruct the route by overestimating the load balancers along the paths toward the destination and (ii) erroneously infer routing changes. Categories and Subject Descriptors C.2.1 [Computer-communication networks]: Network Architec- ture and Design—Network topology (a) Traceroute reports two addresses at the 8-th hop. The common interpretation is that the 7-th hop is splitting the traffic along two Keywords different forwarding paths (case 1); another explanation is that the 8- th hop is an RFC compliant router using multiple interfaces to reply Internet topology; Traceroute; IP alias resolution; IP to Router to the source (case 2). mapping 1 1. INTRODUCTION 0.8 Operators and researchers rely on traceroute to measure routes and they assume that, if traceroute returns different IPs at a given 0.6 hop, it indicates different paths. However, this is not always the case. Although state-of-the-art implementations of traceroute al- 0.4 low to trace all the paths -

Forest Quickstart Guide for Linguists
Forest Quickstart Guide for Linguists Guido Vanden Wyngaerd [email protected] June 28, 2020 Contents 1 Introduction 1 2 Loading Forest 2 3 Basic Usage 2 4 Adjusting node spacing 4 5 Triangles 7 6 Unlabelled nodes 9 7 Horizontal alignment of terminals 10 8 Arrows 11 9 Highlighting 14 1 Introduction Forest is a package for drawing linguistic (and other) tree diagrams de- veloped by Sašo Živanović. This manual provides a quickstart guide for linguists with just the essential things that you need to get started. More 1 extensive documentation is available from the CTAN-archive. Forest is based on the TikZ package; more information about its commands, in par- ticular those controlling the appearance of the nodes, the arrows, and the highlighting can be found in the TikZ documentation. 2 Loading Forest In your preamble, put \usepackage[linguistics]{forest} The linguistics option makes for nice trees, in which the branches meet above the two nodes that they join; it will also align the example number (provided by linguex) with the top of the tree: (1) CP C IP I VP V NP 3 Basic Usage Forest uses a familiar labelled brackets syntax. The code below will out- put the tree in (1) above (\ex. requires the linguex package and provides the example number): \ex. \begin{forest} [CP[C][IP[I][VP[V][NP]]]] \end{forest} Forest will parse the above code without problem, but you are likely to soon get lost in your labelled brackets with more complicated trees if you write the code this way. The better alternative is to arrange the nodes over multiple lines: 2 \ex. -

Problem Solving and Unix Tools
Problem Solving and Unix Tools Command Shell versus Graphical User Interface • Ease of use • Interactive exploration • Scalability • Complexity • Repetition Example: Find all Tex files in a directory (and its subdirectories) that have not changed in the past 21 days. With an interactive file roller, it is easy to sort files by particular characteristics such as the file extension and the date. But this sorting does not apply to files within subdirectories of the current directory, and it is difficult to apply more than one sort criteria at a time. A command line interface allows us to construct a more complex search. In unix, we find the files we are after by executing the command, find /home/nolan/ -mtime +21 -name ’*.tex’ To find out more about a command you can read the online man pages man find or you can execute the command with the –help option. In this example, the standard output to the screen is piped into the more command which formats it to dispaly one screenful at a time. Hitting the space bar displays the next page of output, the return key displays the next line of output, and the ”q” key quits the display. find --help | more Construct Solution in Pieces • Solve a problem by breaking down into pieces and building back up • Typing vs automation • Error messages - experimentation 1 Example: Find all occurrences of a particular string in several files. The grep command searches the contents of files for a regular expression. In this case we search for the simple character string “/stat141/FINAL” in all files in the directory WebLog that begin with the filename “access”. -

What Is UNIX? the Directory Structure Basic Commands Find
What is UNIX? UNIX is an operating system like Windows on our computers. By operating system, we mean the suite of programs which make the computer work. It is a stable, multi-user, multi-tasking system for servers, desktops and laptops. The Directory Structure All the files are grouped together in the directory structure. The file-system is arranged in a hierarchical structure, like an inverted tree. The top of the hierarchy is traditionally called root (written as a slash / ) Basic commands When you first login, your current working directory is your home directory. In UNIX (.) means the current directory and (..) means the parent of the current directory. find command The find command is used to locate files on a Unix or Linux system. find will search any set of directories you specify for files that match the supplied search criteria. The syntax looks like this: find where-to-look criteria what-to-do All arguments to find are optional, and there are defaults for all parts. where-to-look defaults to . (that is, the current working directory), criteria defaults to none (that is, select all files), and what-to-do (known as the find action) defaults to ‑print (that is, display the names of found files to standard output). Examples: find . –name *.txt (finds all the files ending with txt in current directory and subdirectories) find . -mtime 1 (find all the files modified exact 1 day) find . -mtime -1 (find all the files modified less than 1 day) find . -mtime +1 (find all the files modified more than 1 day) find . -

NETSTAT Command
NETSTAT Command | NETSTAT Command | Use the NETSTAT command to display network status of the local host. | | ┌┐────────────── | 55──NETSTAT─────6─┤ Option ├─┴──┬────────────────────────────────── ┬ ─ ─ ─ ────────────────────────────────────────5% | │┌┐───────────────────── │ | └─(──SELect───6─┤ Select_String ├─┴ ─ ┘ | Option: | ┌┐─COnn────── (1, 2) ──────────────── | ├──┼─────────────────────────── ┼ ─ ──────────────────────────────────────────────────────────────────────────────┤ | ├─ALL───(2)──────────────────── ┤ | ├─ALLConn─────(1, 2) ────────────── ┤ | ├─ARp ipaddress───────────── ┤ | ├─CLients─────────────────── ┤ | ├─DEvlinks────────────────── ┤ | ├─Gate───(3)─────────────────── ┤ | ├─┬─Help─ ┬─ ───────────────── ┤ | │└┘─?──── │ | ├─HOme────────────────────── ┤ | │┌┐─2ð────── │ | ├─Interval─────(1, 2) ─┼───────── ┼─ ┤ | │└┘─seconds─ │ | ├─LEVel───────────────────── ┤ | ├─POOLsize────────────────── ┤ | ├─SOCKets─────────────────── ┤ | ├─TCp serverid───(1) ─────────── ┤ | ├─TELnet───(4)───────────────── ┤ | ├─Up──────────────────────── ┤ | └┘─┤ Command ├───(5)──────────── | Command: | ├──┬─CP cp_command───(6) ─ ┬ ────────────────────────────────────────────────────────────────────────────────────────┤ | ├─DELarp ipaddress─ ┤ | ├─DRop conn_num──── ┤ | └─RESETPool──────── ┘ | Select_String: | ├─ ─┬─ipaddress────(3) ┬ ─ ───────────────────────────────────────────────────────────────────────────────────────────┤ | ├─ldev_num─────(4) ┤ | └─userid────(2) ─── ┘ | Notes: | 1 Only ALLCON, CONN and TCP are valid with INTERVAL. | 2 The userid -

Introduction to Unix Shell
Introduction to Unix Shell François Serra, David Castillo, Marc A. Marti- Renom Genome Biology Group (CNAG) Structural Genomics Group (CRG) Run Store Programs Data Communicate Interact with each other with us The Unix Shell Introduction Interact with us Rewiring Telepathy Typewriter Speech WIMP The Unix Shell Introduction user logs in The Unix Shell Introduction user logs in user types command The Unix Shell Introduction user logs in user types command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output ⋮ user logs off shell The Unix Shell Introduction user logs in user types command computer executes command and prints output user types another command computer executes command and prints output -

CS101 Lecture 8
What is a program? What is a “Window Manager” ? What is a “GUI” ? How do you navigate the Unix directory tree? What is a wildcard? Readings: See CCSO’s Unix pages and 8-2 Applications Unix Engineering Workstations UNIX- operating system / C- programming language / Matlab Facilitate machine independent program development Computer program(software): a sequence of instructions that tells the computer what to do. A program takes raw data as input and produces information as output. System Software: • Operating Systems Unix,Windows,MacOS,VM/CMS,... • Editor Programs gedit, pico, vi Applications Software: • Translators and Interpreters gcc--gnu c compiler matlab – interpreter • User created Programs!!! 8-4 X-windows-a Window Manager and GUI(Graphical User Interface) Click Applications and follow the menus or click on an icon to run a program. Click Terminal to produce a command line interface. When the following slides refer to Unix commands it is assumed that these are entered on the command line that begins with the symbol “>” (prompt symbol). Data, information, computer instructions, etc. are saved in secondary storage (auxiliary storage) in files. Files are collected or organized in directories. Each user on a multi-user system has his/her own home directory. In Unix users and system administrators organize files and directories in a hierarchical tree. secondary storage Home Directory When a user first logs in on a computer, the user will be “in” the users home directory. To find the name of the directory type > pwd (this is an acronym for print working directory) So the user is in the directory “gambill” The string “/home/gambill” is called a path. -

Trees, Binary Search Trees, Heaps & Applications Dr. Chris Bourke
Trees Trees, Binary Search Trees, Heaps & Applications Dr. Chris Bourke Department of Computer Science & Engineering University of Nebraska|Lincoln Lincoln, NE 68588, USA [email protected] http://cse.unl.edu/~cbourke 2015/01/31 21:05:31 Abstract These are lecture notes used in CSCE 156 (Computer Science II), CSCE 235 (Dis- crete Structures) and CSCE 310 (Data Structures & Algorithms) at the University of Nebraska|Lincoln. This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License 1 Contents I Trees4 1 Introduction4 2 Definitions & Terminology5 3 Tree Traversal7 3.1 Preorder Traversal................................7 3.2 Inorder Traversal.................................7 3.3 Postorder Traversal................................7 3.4 Breadth-First Search Traversal..........................8 3.5 Implementations & Data Structures.......................8 3.5.1 Preorder Implementations........................8 3.5.2 Inorder Implementation.........................9 3.5.3 Postorder Implementation........................ 10 3.5.4 BFS Implementation........................... 12 3.5.5 Tree Walk Implementations....................... 12 3.6 Operations..................................... 12 4 Binary Search Trees 14 4.1 Basic Operations................................. 15 5 Balanced Binary Search Trees 17 5.1 2-3 Trees...................................... 17 5.2 AVL Trees..................................... 17 5.3 Red-Black Trees.................................. 19 6 Optimal Binary Search Trees 19 7 Heaps 19