Lecture 2: Kinetics: As Described As Transformation Rate Between Two Equilibrium States
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamic Potentials and Natural Variables
Revista Brasileira de Ensino de Física, vol. 42, e20190127 (2020) Articles www.scielo.br/rbef cb DOI: http://dx.doi.org/10.1590/1806-9126-RBEF-2019-0127 Licença Creative Commons Thermodynamic Potentials and Natural Variables M. Amaku1,2, F. A. B. Coutinho*1, L. N. Oliveira3 1Universidade de São Paulo, Faculdade de Medicina, São Paulo, SP, Brasil 2Universidade de São Paulo, Faculdade de Medicina Veterinária e Zootecnia, São Paulo, SP, Brasil 3Universidade de São Paulo, Instituto de Física de São Carlos, São Carlos, SP, Brasil Received on May 30, 2019. Revised on September 13, 2018. Accepted on October 4, 2019. Most books on Thermodynamics explain what thermodynamic potentials are and how conveniently they describe the properties of physical systems. Certain books add that, to be useful, the thermodynamic potentials must be expressed in their “natural variables”. Here we show that, given a set of physical variables, an appropriate thermodynamic potential can always be defined, which contains all the thermodynamic information about the system. We adopt various perspectives to discuss this point, which to the best of our knowledge has not been clearly presented in the literature. Keywords: Thermodynamic Potentials, natural variables, Legendre transforms. 1. Introduction same statement cannot be applied to the temperature. In real fluids, even in simple ones, the proportionality Basic concepts are most easily understood when we dis- to T is washed out, and the Internal Energy is more cuss simple systems. Consider an ideal gas in a cylinder. conveniently expressed as a function of the entropy and The cylinder is closed, its walls are conducting, and a volume: U = U(S, V ). -

PDF Version of Helmholtz Free Energy
Free energy Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS which leads to dU - TdS ≤ 0 Since T and V are constant we can write this as d(U - TS) ≤ 0 The quantity in parentheses is a measure of the spontaneity of the system that depends on known state functions. Definition of Helmholtz Free Energy We define a new state function: A = U -TS such that dA ≤ 0. We call A the Helmholtz free energy. At constant T and V the Helmholtz free energy will decrease until all possible spontaneous processes have occurred. At that point the system will be in equilibrium. The condition for equilibrium is dA = 0. A time Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. The condition for spontaneous change is that ∆A is less than zero and the condition for equilibrium is that ∆A = 0. We write ∆A = ∆U – T∆S ≤ 0 (at constant T and V) Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Article A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action Ivan Kennedy 1,2,*, Harold Geering 2, Michael Rose 3 and Angus Crossan 2 1 Sydney Institute of Agriculture, University of Sydney, NSW 2006, Australia 2 QuickTest Technologies, PO Box 6285 North Ryde, NSW 2113, Australia; [email protected] (H.G.); [email protected] (A.C.) 3 NSW Department of Primary Industries, Wollongbar NSW 2447, Australia; [email protected] * Correspondence: [email protected]; Tel.: + 61-4-0794-9622 Received: 23 March 2019; Accepted: 26 April 2019; Published: 1 May 2019 Abstract: A convenient practical model for accurately estimating the total entropy (ΣSi) of atmospheric gases based on physical action is proposed. This realistic approach is fully consistent with statistical mechanics, but reinterprets its partition functions as measures of translational, rotational, and vibrational action or quantum states, to estimate the entropy. With all kinds of molecular action expressed as logarithmic functions, the total heat required for warming a chemical system from 0 K (ΣSiT) to a given temperature and pressure can be computed, yielding results identical with published experimental third law values of entropy. All thermodynamic properties of gases including entropy, enthalpy, Gibbs energy, and Helmholtz energy are directly estimated using simple algorithms based on simple molecular and physical properties, without resource to tables of standard values; both free energies are measures of quantum field states and of minimal statistical degeneracy, decreasing with temperature and declining density. We propose that this more realistic approach has heuristic value for thermodynamic computation of atmospheric profiles, based on steady state heat flows equilibrating with gravity. -

LECTURES on MATHEMATICAL STATISTICAL MECHANICS S. Adams
ISSN 0070-7414 Sgr´ıbhinn´ıInstiti´uid Ard-L´einnBhaile´ Atha´ Cliath Sraith. A. Uimh 30 Communications of the Dublin Institute for Advanced Studies Series A (Theoretical Physics), No. 30 LECTURES ON MATHEMATICAL STATISTICAL MECHANICS By S. Adams DUBLIN Institi´uid Ard-L´einnBhaile´ Atha´ Cliath Dublin Institute for Advanced Studies 2006 Contents 1 Introduction 1 2 Ergodic theory 2 2.1 Microscopic dynamics and time averages . .2 2.2 Boltzmann's heuristics and ergodic hypothesis . .8 2.3 Formal Response: Birkhoff and von Neumann ergodic theories9 2.4 Microcanonical measure . 13 3 Entropy 16 3.1 Probabilistic view on Boltzmann's entropy . 16 3.2 Shannon's entropy . 17 4 The Gibbs ensembles 20 4.1 The canonical Gibbs ensemble . 20 4.2 The Gibbs paradox . 26 4.3 The grandcanonical ensemble . 27 4.4 The "orthodicity problem" . 31 5 The Thermodynamic limit 33 5.1 Definition . 33 5.2 Thermodynamic function: Free energy . 37 5.3 Equivalence of ensembles . 42 6 Gibbs measures 44 6.1 Definition . 44 6.2 The one-dimensional Ising model . 47 6.3 Symmetry and symmetry breaking . 51 6.4 The Ising ferromagnet in two dimensions . 52 6.5 Extreme Gibbs measures . 57 6.6 Uniqueness . 58 6.7 Ergodicity . 60 7 A variational characterisation of Gibbs measures 62 8 Large deviations theory 68 8.1 Motivation . 68 8.2 Definition . 70 8.3 Some results for Gibbs measures . 72 i 9 Models 73 9.1 Lattice Gases . 74 9.2 Magnetic Models . 75 9.3 Curie-Weiss model . 77 9.4 Continuous Ising model . -

Entropy and H Theorem the Mathematical Legacy of Ludwig Boltzmann
ENTROPY AND H THEOREM THE MATHEMATICAL LEGACY OF LUDWIG BOLTZMANN Cambridge/Newton Institute, 15 November 2010 C´edric Villani University of Lyon & Institut Henri Poincar´e(Paris) Cutting-edge physics at the end of nineteenth century Long-time behavior of a (dilute) classical gas Take many (say 1020) small hard balls, bouncing against each other, in a box Let the gas evolve according to Newton’s equations Prediction by Maxwell and Boltzmann The distribution function is asymptotically Gaussian v 2 f(t, x, v) a exp | | as t ≃ − 2T → ∞ Based on four major conceptual advances 1865-1875 Major modelling advance: Boltzmann equation • Major mathematical advance: the statistical entropy • Major physical advance: macroscopic irreversibility • Major PDE advance: qualitative functional study • Let us review these advances = journey around centennial scientific problems ⇒ The Boltzmann equation Models rarefied gases (Maxwell 1865, Boltzmann 1872) f(t, x, v) : density of particles in (x, v) space at time t f(t, x, v) dxdv = fraction of mass in dxdv The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

A Molecular Modeler's Guide to Statistical Mechanics
A Molecular Modeler’s Guide to Statistical Mechanics Course notes for BIOE575 Daniel A. Beard Department of Bioengineering University of Washington Box 3552255 [email protected] (206) 685 9891 April 11, 2001 Contents 1 Basic Principles and the Microcanonical Ensemble 2 1.1 Classical Laws of Motion . 2 1.2 Ensembles and Thermodynamics . 3 1.2.1 An Ensembles of Particles . 3 1.2.2 Microscopic Thermodynamics . 4 1.2.3 Formalism for Classical Systems . 7 1.3 Example Problem: Classical Ideal Gas . 8 1.4 Example Problem: Quantum Ideal Gas . 10 2 Canonical Ensemble and Equipartition 15 2.1 The Canonical Distribution . 15 2.1.1 A Derivation . 15 2.1.2 Another Derivation . 16 2.1.3 One More Derivation . 17 2.2 More Thermodynamics . 19 2.3 Formalism for Classical Systems . 20 2.4 Equipartition . 20 2.5 Example Problem: Harmonic Oscillators and Blackbody Radiation . 21 2.5.1 Classical Oscillator . 22 2.5.2 Quantum Oscillator . 22 2.5.3 Blackbody Radiation . 23 2.6 Example Application: Poisson-Boltzmann Theory . 24 2.7 Brief Introduction to the Grand Canonical Ensemble . 25 3 Brownian Motion, Fokker-Planck Equations, and the Fluctuation-Dissipation Theo- rem 27 3.1 One-Dimensional Langevin Equation and Fluctuation- Dissipation Theorem . 27 3.2 Fokker-Planck Equation . 29 3.3 Brownian Motion of Several Particles . 30 3.4 Fluctuation-Dissipation and Brownian Dynamics . 32 1 Chapter 1 Basic Principles and the Microcanonical Ensemble The first part of this course will consist of an introduction to the basic principles of statistical mechanics (or statistical physics) which is the set of theoretical techniques used to understand microscopic systems and how microscopic behavior is reflected on the macroscopic scale. -

Thermodynamic Analysis Based on the Second-Order Variations of Thermodynamic Potentials
Theoret. Appl. Mech., Vol.35, No.1-3, pp. 215{234, Belgrade 2008 Thermodynamic analysis based on the second-order variations of thermodynamic potentials Vlado A. Lubarda ¤ Abstract An analysis of the Gibbs conditions of stable thermodynamic equi- librium, based on the constrained minimization of the four fundamen- tal thermodynamic potentials, is presented with a particular attention given to the previously unexplored connections between the second- order variations of thermodynamic potentials. These connections are used to establish the convexity properties of all potentials in relation to each other, which systematically deliver thermodynamic relationships between the speci¯c heats, and the isentropic and isothermal bulk mod- uli and compressibilities. The comparison with the classical derivation is then given. Keywords: Gibbs conditions, internal energy, second-order variations, speci¯c heats, thermodynamic potentials 1 Introduction The Gibbs conditions of thermodynamic equilibrium are of great importance in the analysis of the equilibrium and stability of homogeneous and heterogeneous thermodynamic systems [1]. The system is in a thermodynamic equilibrium if its state variables do not spontaneously change with time. As a consequence of the second law of thermodynamics, the equilibrium state of an isolated sys- tem at constant volume and internal energy is the state with the maximum value of the total entropy. Alternatively, among all neighboring states with the ¤Department of Mechanical and Aerospace Engineering, University of California, San Diego; La Jolla, CA 92093-0411, USA and Montenegrin Academy of Sciences and Arts, Rista Stijovi¶ca5, 81000 Podgorica, Montenegro, e-mail: [email protected]; [email protected] 215 216 Vlado A. Lubarda same volume and total entropy, the equilibrium state is one with the lowest total internal energy. -

Entropy: from the Boltzmann Equation to the Maxwell Boltzmann Distribution
Entropy: From the Boltzmann equation to the Maxwell Boltzmann distribution A formula to relate entropy to probability Often it is a lot more useful to think about entropy in terms of the probability with which different states are occupied. Lets see if we can describe entropy as a function of the probability distribution between different states. N! WN particles = n1!n2!....nt! stirling (N e)N N N W = = N particles n1 n2 nt n1 n2 nt (n1 e) (n2 e) ....(nt e) n1 n2 ...nt with N pi = ni 1 W = N particles n1 n2 nt p1 p2 ...pt takeln t lnWN particles = "#ni ln pi i=1 divide N particles t lnW1particle = "# pi ln pi i=1 times k t k lnW1particle = "k# pi ln pi = S1particle i=1 and t t S = "N k p ln p = "R p ln p NA A # i # i i i=1 i=1 ! Think about how this equation behaves for a moment. If any one of the states has a probability of occurring equal to 1, then the ln pi of that state is 0 and the probability of all the other states has to be 0 (the sum over all probabilities has to be 1). So the entropy of such a system is 0. This is exactly what we would have expected. In our coin flipping case there was only one way to be all heads and the W of that configuration was 1. Also, and this is not so obvious, the maximal entropy will result if all states are equally populated (if you want to see a mathematical proof of this check out Ken Dill’s book on page 85). -
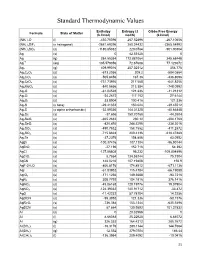
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

Boltzmann Equation II: Binary Collisions
Physics 127b: Statistical Mechanics Boltzmann Equation II: Binary Collisions Binary collisions in a classical gas Scattering out Scattering in v'1 v'2 v 1 v 2 R v 2 v 1 v'2 v'1 Center of V' Mass Frame V θ θ b sc sc R V V' Figure 1: Binary collisions in a gas: top—lab frame; bottom—centre of mass frame Binary collisions in a gas are very similar, except that the scattering is off another molecule. An individual scattering process is, of course, simplest to describe in the center of mass frame in terms of the relative E velocity V =Ev1 −Ev2. However the center of mass frame is different for different collisions, so we must keep track of the results in the lab frame, and this makes the calculation rather intricate. I will indicate the main ideas here, and refer you to Reif or Landau and Lifshitz for precise discussions. Lets first set things up in the lab frame. Again we consider the pair of scattering in and scattering out processes that are space-time inverses, and so have identical cross sections. We can write abstractly for the scattering out from velocity vE1 due to collisions with molecules with all velocities vE2, which will clearly be proportional to the number f (vE1) of molecules at vE1 (which we write as f1—sorry, not the same notation as in the previous sections where f1 denoted the deviation of f from the equilibrium distribution!) and the 3 3 number f2d v2 ≡ f(vE2)d v2 in each velocity volume element, and then we must integrate over all possible vE0 vE0 outgoing velocities 1 and 2 ZZZ df (vE ) 1 =− w(vE0 , vE0 ;Ev , vE )f f d3v d3v0 d3v0 .