Gaba Conjugates and Methods of Use Thereof Gaba-Konjugate Und Verfahren Zu Ihrer Verwendung Conjugués Gaba Et Procédés D’Utilisation De Ceux-Ci
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Oral Presentations September 23Rd - Rooms 1,2 and 3
Oral Presentations September 23rd - Rooms 1,2 and 3 Presentation Date Abstract Authors Presenter´s name - Theme Title Code indicated by the author 18498 Thomas Smits; Femke Gresnigt; Thomas Smits Clinical Toxicology/drugs of PERFORMANCE OF AN IMMUNOASSAY Eric Franssen; Milly Attema-de abuse METHOD FOR GAMMA-HYDROXYBUTYRIC Jonge ACID (GHB) IN PATIENTS PRESENTED AT THE EMERGENCY DEPARTMENT, A PROSPECTIVE STUDY 18499 Thomas Smits; Femke Gresnigt; Thomas Smits Clinical Toxicology/drugs of DO WE NEED POINT-OF-CARE TESTING OF Milly Attema-de Jonge; Eric abuse GAMMA-HYDROXYBUTYRIC ACID (GHB) AT Fransse THE EMERGENCY DEPARTMENT? September 23 18730 Lilian H.J. Richter; Julia Menges; Lea Wagmann Clinical Toxicology/drugs of NEW PSYCHOACTIVE SUBSTANCES: Lea Wagmann; Simon D. Brandt; abuse METABOLIC FATE, ISOZYME-MAPPING, 13:30 - 14:45 Folker Westphal; Veit Flockerzi; AND PLASMA PROTEIN BINDING OF 5-APB- ROOM 1 Markus R. Meyer NBOME, 2C-B-FLY-NB2ETO5CL, AND 2C-B- FLY-NBOME 18985 Annelies Cannaert; Marie Annelies Cannaert Clinical Toxicology/drugs of HIDE AND SEEK: OVERCOMING THE Deventer; Melissa Fogarty; abuse MASKING EFFECT OF OPIOID Amanda L.A. Mohr; Christophe P. ANTAGONISTS IN ACTIVITY-BASED Stove SCREENING TESTS 18740 Souleiman El Balkhi ; Roland Souleiman El Balkhi Clinical Toxicology/drugs of METABOLIC INTERACTIONS BETWEEN Lawson; Franck Saint-Marcoux abuse OXYCODONE, BENZODIAZEPINES OR DESIGNER BENZODIAZEPINES PLAY AN IMPORTANT ROLE IN OXYCODONE INTOXICATIONS 19050 Brenda de Winter F de Velde; MN Brenda de Winter Anti-infective drugs POPULATION -

Summary: Traditionally, Bioanalytical Laboratories Do Not Report Actual
Summary: Traditionally, bioanalytical laboratories do not report actual concentrations for samples with results below the limit of quantification (BLQ) in pharmacokinetic studies. BLQ values are outside the method calibration range established during validation and no data are available to support the reliability of these values. However, ignoring BLQ data can contribute to bias and imprecision in model-based pharmacokinetic analyses. From this perspective, routine use of BLQ data would be advantageous. We would like to initiate an interdisciplinary debate on this important topic by summarising the current concepts and use of BLQ data by regulators, pharmacometricians and bioanalysts. Through introducing the limit of detection and evaluating its variability BLQ data could be released and utilized appropriately for pharmacokinetic research. Keywords: Lower limit of quantification (LLOQ) Below limit of quantification (BLQ) result Limit of detection (LoD) Pharmacokinetic (PK) Pharmacodynamics (PD) Introduction Studying the effects of drugs remains central to both medical research and clinical practice. Two key branches of pharmacological analysis are (i) pharmacokinetics (PK), including drug absorption, distribution, metabolism and elimination, and (ii) pharmacodynamics (PD), exploring the effects of drugs on the living organism, including efficacy and toxicity. In PK studies the samples are collected in an effort to map the drug concentration over time in the patient. For samples collected many hours post-dose drug concentrations may be low, yet can still provide valuable information on pharmacokinetic parameters such as clearance [1,2]. Similarly, in the case of biomarker PD studies, concentrations that are too low to quantify with a particular bioanalytical method may still provide useful information. Bioanalytical laboratories define the lowest concentration that can be quantified accurately by a method as the lower limit of quantification (LLOQ). -
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
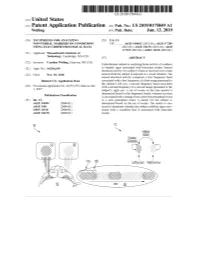
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

Chapter 25 Mechanisms of Action of Antiepileptic Drugs
Chapter 25 Mechanisms of action of antiepileptic drugs GRAEME J. SILLS Department of Molecular and Clinical Pharmacology, University of Liverpool _________________________________________________________________________ Introduction The serendipitous discovery of the anticonvulsant properties of phenobarbital in 1912 marked the foundation of the modern pharmacotherapy of epilepsy. The subsequent 70 years saw the introduction of phenytoin, ethosuximide, carbamazepine, sodium valproate and a range of benzodiazepines. Collectively, these compounds have come to be regarded as the ‘established’ antiepileptic drugs (AEDs). A concerted period of development of drugs for epilepsy throughout the 1980s and 1990s has resulted (to date) in 16 new agents being licensed as add-on treatment for difficult-to-control adult and/or paediatric epilepsy, with some becoming available as monotherapy for newly diagnosed patients. Together, these have become known as the ‘modern’ AEDs. Throughout this period of unprecedented drug development, there have also been considerable advances in our understanding of how antiepileptic agents exert their effects at the cellular level. AEDs are neither preventive nor curative and are employed solely as a means of controlling symptoms (i.e. suppression of seizures). Recurrent seizure activity is the manifestation of an intermittent and excessive hyperexcitability of the nervous system and, while the pharmacological minutiae of currently marketed AEDs remain to be completely unravelled, these agents essentially redress the balance between neuronal excitation and inhibition. Three major classes of mechanism are recognised: modulation of voltage-gated ion channels; enhancement of gamma-aminobutyric acid (GABA)-mediated inhibitory neurotransmission; and attenuation of glutamate-mediated excitatory neurotransmission. The principal pharmacological targets of currently available AEDs are highlighted in Table 1 and discussed further below. -

“GABA” 'Bout? Pregabalin and Gabapentin Abuse
3/19/18 What’s All the “GABA” ‘Bout? Pregabalin and Gabapentin Abuse Courtney Kominek, PharmD, BCPS, CPE Disclosures .Courtney Kominek, PharmD, BCPS, CPE –Axial Healthcare – Consultant .The views and opinions expressed in this presentation are those of the authors and do not necessarily reflect the official policy or position of any agency of the United States government, including the Department of Veterans Affairs. 1 3/19/18 Learning Objectives .Review the proposed mechanisms of action (MOA) for gabapentin and pregabalin. .Explain the proposed rationale as to why gabapentin and pregabalin have become drugs of abuse. .Identify signs and symptoms of withdrawal that an addicted or tolerant patient may experience upon abrupt discontinuation of gabapentin or pregabalin. .Discuss updates on changes in pain management given the increase in gabapentin and pregabalin abuse. Current Situation Opioid overdose public health crisis Rising use of nonopioid medications including gabapentin Opioids and concomitant gabapentin increase risk for overdose Reports of gabapentinoid abuse Changes in PDMP and scheduling at state level http://www.register-herald.com/news/manchin-asks-fda-dea-to-consider-rescheduling-gabapentin/article_442fa04b-7ed9-5bf8-8d19-b5440e9c278b.html 2 3/19/18 Gabapentin and Pregabalin: Pharmacology and Pharmacokinetics Fact or Alternate Fact? .Gabapentin and pregabalin work on GABA. 3 3/19/18 Mechanism of Action Structurally related to GABA and has GABA-mimetic properties Do not • Alter uptake or breakdown • Convert into GABA • Bind to GABAa or GABAB Binds to the α2-δ subunit of the voltage-gated calcium channel Reduces the Ca2+ -dependent release of pro-nociceptive neurotransmitters Decreases release of glutamate, NE, and substance P Dworkin RH et al. -

5-HT2A/2C Receptor Modulation of Absence Seizures and Characterization of the GHB-Model
5-HT2A/2C receptor modulation of absence seizures and characterization of the GHB-model A thesis submitted to Cardiff University for the degree of Doctor of Philosophy by Marcello Venzi (MSc) School of Biosciences, Cardiff University, October 2014 Chapter 1 Declaration and Statements This work has not been submitted in substance for any other degree or award at this or any other university or place of learning, nor is being submitted concurrently in candidature for any degree or other award. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 1 This thesis is being submitted in partial fulfillment of the requirements for the degree of PhD . Signed ………………………………………… (candidate) Date ………………………… STATEMENT 2 This thesis is the result of my own independent work/investigation, except where otherwise stated. Other sources are acknowledged by explicit references. The views expressed are my own. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 3 I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loan, and for the title and summary to be made available to outside organisations. Signed ………………………………………… (candidate) Date ………………………… STATEMENT 4: PREVIOUSLY APPROVED BAR ON ACCESS I hereby give consent for my thesis, if accepted, to be available for photocopying and for inter- library loans after expiry of a bar on access previously approved by the Academic Standards & Quality Committee. Signed ………………………………………… (candidate) Date ………………………… II Chapter 1 Summary Absence seizures (ASs) are non-convulsive epileptic events which are common in pediatric and juvenile epilepsies. They consist of EEG generalized spike-and-wave-discharges (SWDs) accompanied by an impairment of consciousness and are expressed within the thalamocortical network. -

OCUFLOX (Ofloxacin Ophthalmic Solution) 0.3% Sterile
® OCUFLOX (ofloxacin ophthalmic solution) 0.3% sterile DESCRIPTION OCUFLOX® (ofloxacin ophthalmic solution) 0.3% is a sterile ophthalmic solution. It is a fluorinated carboxyquinolone anti-infective for topical ophthalmic use. Chemical Name: (±)-9-Fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyrido[1,2,3-de]-1,4-benzoxazine-6- carboxylic acid. Contains: Active: ofloxacin 0.3% (3 mg/mL). Preservative: benzalkonium chloride (0.005%). Inactives: sodium chloride and purified water. May also contain hydrochloric acid and/or sodium hydroxide to adjust pH. OCUFLOX® solution is unbuffered and formulated with a pH of 6.4 (range - 6.0 to 6.8). It has an osmolality of 300 mOsm/kg. Ofloxacin is a fluorinated 4-quinolone which differs from other fluorinated 4-quinolones in that there is a six member (pyridobenzoxazine) ring from positions 1 to 8 of the basic ring structure. CLINICAL PHARMACOLOGY Pharmacokinetics Serum, urine and tear concentrations of ofloxacin were measured in 30 healthy women at various time points during a ten-day course of treatment with OCUFLOX® solution. The mean serum ofloxacin concentration ranged from 0.4 ng/mL to 1.9 ng/mL. Maximum ofloxacin concentration increased from 1.1 ng/mL on day one to 1.9 ng/mL on day 11 after QID dosing for 10 1/2 days. Maximum serum ofloxacin concentrations after ten days of topical ophthalmic dosing were more than 1000 times lower than those reported after standard oral doses of ofloxacin. Tear ofloxacin concentrations ranged from 5.7 to 31 mcg/g during the 40 minute period following the last dose on day 11. -

Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review
Current Pain and Headache Reports (2019) 23: 37 https://doi.org/10.1007/s11916-019-0774-0 OTHER PAIN (A KAYE AND N VADIVELU, SECTION EDITORS) Membrane Stabilizer Medications in the Treatment of Chronic Neuropathic Pain: a Comprehensive Review Omar Viswanath1,2,3 & Ivan Urits4 & Mark R. Jones4 & Jacqueline M. Peck5 & Justin Kochanski6 & Morgan Hasegawa6 & Best Anyama7 & Alan D. Kaye7 Published online: 1 May 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract Purpose of Review Neuropathic pain is often debilitating, severely limiting the daily lives of patients who are affected. Typically, neuropathic pain is difficult to manage and, as a result, leads to progression into a chronic condition that is, in many instances, refractory to medical management. Recent Findings Gabapentinoids, belonging to the calcium channel blocking class of drugs, have shown good efficacy in the management of chronic pain and are thus commonly utilized as first-line therapy. Various sodium channel blocking drugs, belonging to the categories of anticonvulsants and local anesthetics, have demonstrated varying degrees of efficacy in the in the treatment of neurogenic pain. Summary Though there is limited medical literature as to efficacy of any one drug, individualized multimodal therapy can provide significant analgesia to patients with chronic neuropathic pain. Keywords Neuropathic pain . Chronic pain . Ion Channel blockers . Anticonvulsants . Membrane stabilizers Introduction Neuropathic pain, which is a result of nervous system injury or lives of patients who are affected. Frequently, it is difficult to dysfunction, is often debilitating, severely limiting the daily manage and as a result leads to the progression of a chronic condition that is, in many instances, refractory to medical This article is part of the Topical Collection on Other Pain management. -

Case 2:14-Cv-05824-ES-JAD Document 1 Filed 09/18/14 Page 1 of 35 Pageid: 1
Case 2:14-cv-05824-ES-JAD Document 1 Filed 09/18/14 Page 1 of 35 PageID: 1 Charles M. Lizza William C. Baton SAUL EWING LLP One Riverfront Plaza, Suite 1520 Newark, New Jersey 07102-5426 (973) 286-6700 [email protected] Attorneys for Plaintiff Jazz Pharmaceuticals, Inc. UNITED STATES DISTRICT COURT DISTRICT OF NEW JERSEY JAZZ PHARMACEUTICALS, INC., Civil Action No. ____________________ Plaintiff, COMPLAINT FOR PATENT INFRINGEMENT v. PAR PHARMACEUTICAL, INC., (Filed Electronically) Defendant. Plaintiff Jazz Pharmaceuticals, Inc. (“Jazz Pharmaceuticals”), by its undersigned attorneys, for its Complaint against defendant Par Pharmaceutical, Inc. (“Par”), alleges as follows: Nature of the Action 1. This is an action for patent infringement under the patent laws of the United States, 35 U.S.C. §100, et seq., arising from Par’s filing of an Abbreviated New Drug Application (“ANDA”) with the United States Food and Drug Administration (“FDA”) seeking ® approval to commercially market a generic version of Jazz Pharmaceuticals’ XYREM drug product prior to the expiration of United States Patent No. 8,772,306 (“the ’306 patent” or “the patent-in-suit”) owned by Jazz Pharmaceuticals. Case 2:14-cv-05824-ES-JAD Document 1 Filed 09/18/14 Page 2 of 35 PageID: 2 The Parties 2. Plaintiff Jazz Pharmaceuticals is a corporation organized and existing under the laws of the State of Delaware, having a principal place of business at 3180 Porter Drive, Palo Alto, California 94304. 3. On information and belief, defendant Par Pharmaceutical, Inc. is a corporation organized and existing under the laws of the State of Delaware, having a principal place of business at 300 Tice Boulevard, Woodcliff Lake, New Jersey. -

Pregabalin for the Treatment of Drug and Alcohol Withdrawal Symptoms: a Comprehensive Review
CNS Drugs (2016) 30:1191–1200 DOI 10.1007/s40263-016-0390-z REVIEW ARTICLE Pregabalin for the Treatment of Drug and Alcohol Withdrawal Symptoms: A Comprehensive Review 1 2,3 4 5 Rainer Freynhagen • Miroslav Backonja • Stephan Schug • Gavin Lyndon • 6 6 6 Bruce Parsons • Stephen Watt • Regina Behar Published online: 16 November 2016 Ó The Author(s) 2016. This article is published with open access at Springerlink.com Abstract Treatments for physical dependence and asso- treatment of physical dependence and accompanying ciated withdrawal symptoms following the abrupt discon- withdrawal symptoms associated with opioids, benzodi- tinuation of prescription drugs (such as opioids and azepines, nicotine, cannabinoids, and alcohol, although benzodiazepines), nicotine, alcohol, and cannabinoids are data from randomized controlled studies are sparse. How- available, but there is still a need for new and more ever, the current evidence is promising and provides a effective therapies. This review examines evidence sup- platform for future studies, including appropriate random- porting the potential use of pregabalin, an a2d voltage- ized, placebo- and/or comparator-controlled studies, to gated calcium channel subunit ligand, for the treatment of further explore the efficacy and safety of pregabalin for the physical dependence and associated withdrawal symptoms. treatment of withdrawal symptoms. Given the potential for A literature search of the MEDLINE and Cochrane Library pregabalin misuse or abuse, particularly in individuals with databases up to and including 11 December 2015 was a previous history of substance abuse, clinicians should conducted. The search term used was ‘(dependence OR exercise caution when using pregabalin in this patient withdrawal) AND pregabalin’. -

Swedres-Svarm 2019
2019 SWEDRES|SVARM Sales of antibiotics and occurrence of antibiotic resistance in Sweden 2 SWEDRES |SVARM 2019 A report on Swedish Antibiotic Sales and Resistance in Human Medicine (Swedres) and Swedish Veterinary Antibiotic Resistance Monitoring (Svarm) Published by: Public Health Agency of Sweden and National Veterinary Institute Editors: Olov Aspevall and Vendela Wiener, Public Health Agency of Sweden Oskar Nilsson and Märit Pringle, National Veterinary Institute Addresses: The Public Health Agency of Sweden Solna. SE-171 82 Solna, Sweden Östersund. Box 505, SE-831 26 Östersund, Sweden Phone: +46 (0) 10 205 20 00 Fax: +46 (0) 8 32 83 30 E-mail: [email protected] www.folkhalsomyndigheten.se National Veterinary Institute SE-751 89 Uppsala, Sweden Phone: +46 (0) 18 67 40 00 Fax: +46 (0) 18 30 91 62 E-mail: [email protected] www.sva.se Text, tables and figures may be cited and reprinted only with reference to this report. Images, photographs and illustrations are protected by copyright. Suggested citation: Swedres-Svarm 2019. Sales of antibiotics and occurrence of resistance in Sweden. Solna/Uppsala ISSN1650-6332 ISSN 1650-6332 Article no. 19088 This title and previous Swedres and Svarm reports are available for downloading at www.folkhalsomyndigheten.se/ Scan the QR code to open Swedres-Svarm 2019 as a pdf in publicerat-material/ or at www.sva.se/swedres-svarm/ your mobile device, for reading and sharing. Use the camera in you’re mobile device or download a free Layout: Dsign Grafisk Form, Helen Eriksson AB QR code reader such as i-nigma in the App Store for Apple Print: Taberg Media Group, Taberg 2020 devices or in Google Play. -

Penetration of 0.3% Ciprofloxacin, 0.3% Ofloxacin, and 0.5
Brazilian Journal of Medical and Biological Research (2017) 50(7): e5901, http://dx.doi.org/10.1590/1414-431X20175901 ISSN 1414-431X 1/6 Penetration of 0.3% ciprofloxacin, 0.3% ofloxacin, and 0.5% moxifloxacin into the cornea and aqueous humor of enucleated human eyes G.C.M. Silva1, V.A.P. Jabor2, P.S. Bonato2, E.Z. Martinez3 and S.J. Faria-e-Sousa1 1Departamento de Oftalmologia, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil 2Departamento de Física e Química, Faculdade de Ciências Farmacêuticas de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil 3Departamento de Medicina Social, Faculdade de Medicina de Ribeirão Preto, Universidade de São Paulo, Ribeirão Preto, SP, Brasil Abstract We aimed to quantify the penetration of ciprofloxacin, ofloxacin, and moxifloxacin into the cornea and aqueous humor of cadaver eyes. A total of 60 enucleated eyes, not eligible for corneal transplantation, were divided into three groups and immersed in commercial solutions of 0.3% ciprofloxacin, 0.3% ofloxacin, or 0.5% moxifloxacin for 10 min. Whole corneas and samples of aqueous humor were then harvested and frozen, and drug concentrations analyzed by liquid chromatography tandem mass spectrometry. The mean corneal concentration of moxifloxacin was twice as high as ofloxacin, and the latter was twice as high as ciprofloxacin. The mean concentration of moxifloxacin in the aqueous humor was four times higher than the other antibiotics, and the mean concentrations of ciprofloxacin and ofloxacin were statistically similar. The amount of drug that penetrated the anterior chamber after a 10-min immersion was far below the safe limit of endothelial toxicity of each preparation.