Characterization and Prediction of ISRE Binding Patterns Across Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cellular Responses to Erbb-2 Overexpression in Human Mammary Luminal Epithelial Cells: Comparison of Mrna and Protein Expression
British Journal of Cancer (2004) 90, 173 – 181 & 2004 Cancer Research UK All rights reserved 0007 – 0920/04 $25.00 www.bjcancer.com Cellular responses to ErbB-2 overexpression in human mammary luminal epithelial cells: comparison of mRNA and protein expression SL White1, S Gharbi1, MF Bertani1, H-L Chan1, MD Waterfield1 and JF Timms*,1 1 Ludwig Institute for Cancer Research, Wing 1.1, Cruciform Building, Gower Street, London WCIE 6BT, UK Microarray analysis offers a powerful tool for studying the mechanisms of cellular transformation, although the correlation between mRNA and protein expression is largely unknown. In this study, a microarray analysis was performed to compare transcription in response to overexpression of the ErbB-2 receptor tyrosine kinase in a model mammary luminal epithelial cell system, and in response to the ErbB-specific growth factor heregulin b1. We sought to validate mRNA changes by monitoring changes at the protein level using a parallel proteomics strategy, and report a surprisingly high correlation between transcription and translation for the subset of genes studied. We further characterised the identified targets and relate differential expression to changes in the biological properties of ErbB-2-overexpressing cells. We found differential regulation of several key cell cycle modulators, including cyclin D2, and downregulation of a large number of interferon-inducible genes, consistent with increased proliferation of the ErbB-2- overexpressing cells. Furthermore, differential expression of genes involved in extracellular matrix modelling and cellular adhesion was linked to altered adhesion of these cells. Finally, we provide evidence for enhanced autocrine activation of MAPK signalling and the AP-1 transcription complex. -
Primepcr™Assay Validation Report
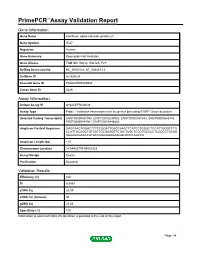
PrimePCR™Assay Validation Report Gene Information Gene Name interferon, alpha-inducible protein 27 Gene Symbol IFI27 Organism Human Gene Summary Description Not Available Gene Aliases FAM14D, ISG12, ISG12A, P27 RefSeq Accession No. NC_000014.8, NT_026437.12 UniGene ID Hs.532634 Ensembl Gene ID ENSG00000165949 Entrez Gene ID 3429 Assay Information Unique Assay ID qHsaCEP0024638 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000557098, ENST00000298902, ENST00000557634, ENST00000555744, ENST00000444961, ENST00000448882 Amplicon Context Sequence GAGCAACTGGACTCTCCGGATTGACCAAGTTCATCCTGGGCTCCATTGGGTCTG CCATTGCGGCTGTCATTGCGAGGTTCTACTAGCTCCCTGCCCCTCGCCCTGCAG AGAAGAGAACCATGCCAGGGGAGAAGGCACCCAGCCA Amplicon Length (bp) 115 Chromosome Location 14:94582779-94582923 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 23.09 cDNA Tm (Celsius) 86 gDNA Cq 23.06 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report IFI27, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus
Gao et al. Hereditas (2021) 158:3 https://doi.org/10.1186/s41065-020-00167-5 RESEARCH Open Access IFI27 may predict and evaluate the severity of respiratory syncytial virus infection in preterm infants Junyan Gao1, Xueping Zhu2, Mingfu Wu1, Lijun Jiang1, Fudong Wang1 and Shan He2,3* Abstract Background: Preterm infants are a special population that vulnerable to respiratory syncytial virus (RSV) infection and the lower respiratory tract infections (LRTIs) caused by RSV could be severe and even life-threating. The purpose of the present study was to identify candidate genes of preterm infants who are susceptible to RSV infection and provide a new insight into the pathogenesis of RSV infection. Methods: Three datasets (GSE77087, GSE69606 and GSE41374) containing 183 blood samples of RSV infected patients and 33 blood samples of healthy controls from Gene Expression Omnibus (GEO) database were downloaded and the differentially expressed genes (DEGs) were screened out. The function and pathway enrichments were analyzed through Database for Annotation, Visualization and Integrated Discovery (DAVID) website. The protein-protein interaction (PPI) network for DEGs was constructed through Search Tool for the Retrieval of Interacting Genes (STRING). The module analysis was performed by Cytoscape software and hub genes were identified. Clinical verification was employed to verify the expression level of top five hub genes among 72 infants including 50 RSV infected patients and 22 non-RSV-infected patients hospitalized in our center. Further, the RSV infected infants with high-expression IFI27 and those with low-expression IFI27 were compared (defined as higher or lower than the median mRNA level). -

IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus Infection in Preterm Infants
IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus Infection in Preterm Infants Junyan Gao Aliated Hospital of Yangzhou University https://orcid.org/0000-0001-6678-6147 Xueping Zhu Children's Hospital of Soochow University Mingfu Wu Aliated Hospital of Yangzhou University Lijun Jiang Aliated Hospital of Yangzhou University Fudong Wang Aliated Hospital of Yangzhou University Shan He ( [email protected] ) Research Keywords: respiratory syncytial virus, preterm infants, differentially expressed genes, protein‐protein interaction, pathway enrichment, interferon alpha inducible protein 27 Posted Date: July 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-42880/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on January 2nd, 2021. See the published version at https://doi.org/10.1186/s41065-020-00167-5. Page 1/21 Abstract Background: Preterm infants are a special population that vulnerable to respiratory syncytial virus (RSV) infection and the lower respiratory tract infection (LRTIs) caused by RSV could be severe and even life- threating. The purpose of the present study was to identify candidate genes of preterm infants who are susceptible to RSV infection and provide a new insight into the pathogenesis of RSV infection. Methods: Three datasets (GSE77087, GSE69606 and GSE41374) containing 183 blood samples of RSV infected patients and 33 blood samples of healthy controls from Gene Expression Omnibus (GEO) database were downloaded and the differentially expressed genes (DEGs) were screened out. The function and pathway enrichments were analyzed through Database for Annotation, Visualization and Integrated Discovery (DAVID) website. -

Integration of 198 Chip-Seq Datasets Reveals Human Cis-Regulatory Regions
Integration of 198 ChIP-seq datasets reveals human cis-regulatory regions Hamid Bolouri & Larry Ruzzo Thanks to Stephen Tapscott, Steven Henikoff & Zizhen Yao Slides will be available from: http://labs.fhcrc.org/bolouri/ Email [email protected] for manuscript (Bolouri & Ruzzo, submitted) Kleinjan & van Heyningen, Am. J. Hum. Genet., 2005, (76)8–32 Epstein, Briefings in Func. Genom. & Protemoics, 2009, 8(4)310-16 Regulation of SPi1 (Sfpi1, PU.1 protein) expression – part 1 miR155*, miR569# ~750nt promoter ~250nt promoter The antisense RNA • causes translational stalling • has its own promoter • requires distal SPI1 enhancer • is transcribed with/without SPI1. # Hikami et al, Arthritis & Rheumatism, 2011, 63(3):755–763 * Vigorito et al, 2007, Immunity 27, 847–859 Ebralidze et al, Genes & Development, 2008, 22: 2085-2092. Regulation of SPi1 expression – part 2 (mouse coordinates) Bidirectional ncRNA transcription proportional to PU.1 expression PU.1/ELF1/FLI1/GLI1 GATA1 GATA1 Sox4/TCF/LEF PU.1 RUNX1 SP1 RUNX1 RUNX1 SP1 ELF1 NF-kB SATB1 IKAROS PU.1 cJun/CEBP OCT1 cJun/CEBP 500b 500b 500b 500b 500b 750b 500b -18Kb -14Kb -12Kb -10Kb -9Kb Chou et al, Blood, 2009, 114: 983-994 Hoogenkamp et al, Molecular & Cellular Biology, 2007, 27(21):7425-7438 Zarnegar & Rothenberg, 2010, Mol. & cell Biol. 4922-4939 An NF-kB binding-site variant in the SPI1 URE reduces PU.1 expression & is GGGCCTCCCC correlated with AML GGGTCTTCCC Bonadies et al, Oncogene, 2009, 29(7):1062-72. SATB1 binding site A distal single nucleotide polymorphism alters long- range regulation of the PU.1 gene in acute myeloid leukemia Steidl et al, J Clin Invest. -

Identification of Key Genes and Pathways in Pancreatic Cancer
G C A T T A C G G C A T genes Article Identification of Key Genes and Pathways in Pancreatic Cancer Gene Expression Profile by Integrative Analysis Wenzong Lu * , Ning Li and Fuyuan Liao Department of Biomedical Engineering, College of Electronic and Information Engineering, Xi’an Technological University, Xi’an 710021, China * Correspondence: [email protected]; Tel.: +86-29-86173358 Received: 6 July 2019; Accepted: 7 August 2019; Published: 13 August 2019 Abstract: Background: Pancreatic cancer is one of the malignant tumors that threaten human health. Methods: The gene expression profiles of GSE15471, GSE19650, GSE32676 and GSE71989 were downloaded from the gene expression omnibus database including pancreatic cancer and normal samples. The differentially expressed genes between the two types of samples were identified with the Limma package using R language. The gene ontology functional and pathway enrichment analyses of differentially-expressed genes were performed by the DAVID software followed by the construction of a protein–protein interaction network. Hub gene identification was performed by the plug-in cytoHubba in cytoscape software, and the reliability and survival analysis of hub genes was carried out in The Cancer Genome Atlas gene expression data. Results: The 138 differentially expressed genes were significantly enriched in biological processes including cell migration, cell adhesion and several pathways, mainly associated with extracellular matrix-receptor interaction and focal adhesion pathway in pancreatic cancer. The top hub genes, namely thrombospondin 1, DNA topoisomerase II alpha, syndecan 1, maternal embryonic leucine zipper kinase and proto-oncogene receptor tyrosine kinase Met were identified from the protein–protein interaction network. -

Identifying Robust Biomarkers of Infection Through an Omics-Based
medRxiv preprint doi: https://doi.org/10.1101/2020.07.28.20163329; this version posted July 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . 1 Identifying robust biomarkers of infection 2 through an omics-based meta-analysis 3 4 Ashleigh C Myall1,2, Simon Perkins1, David Rushton4, Jonathan David5, Phillippa Spencer6, 5 Andrew R Jones1,&, & Philipp Antczak1,3,&* 6 1Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, 7 United Kingdom 8 2Department of Mathematics, Imperial College London, London, United Kingdom 9 3Center for Molecular Medicine, University of Cologne, Cologne, Germany 10 4Defence and Security Analysis Division, Defence Science and Technology laboratory 11 (DSTL), Porton Down Salisbury, United Kingdom 12 5Chemical, Biological and Radiological Division, Defence Science and Technology 13 laboratory (DSTL), Porton Down Salisbury, United Kingdom 14 6Cyber and Information Systems Division, Defence Science and Technology laboratory 15 (DSTL), Porton Down, Salisbury, United Kingdom 16 17 *Corresponding author 18 E-mail: [email protected] (PA) 19 &Joint Senior Authors 1 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.07.28.20163329; this version posted July 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

The MER41 Family of Hervs Is Uniquely Involved in the Immune-Mediated Regulation of Cognition/Behavior-Related Genes
bioRxiv preprint doi: https://doi.org/10.1101/434209; this version posted October 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The MER41 family of HERVs is uniquely involved in the immune-mediated regulation of cognition/behavior-related genes: pathophysiological implications for autism spectrum disorders Serge Nataf*1, 2, 3, Juan Uriagereka4 and Antonio Benitez-Burraco 5 1CarMeN Laboratory, INSERM U1060, INRA U1397, INSA de Lyon, Lyon-Sud Faculty of Medicine, University of Lyon, Pierre-Bénite, France. 2 University of Lyon 1, Lyon, France. 3Banque de Tissus et de Cellules des Hospices Civils de Lyon, Hôpital Edouard Herriot, Lyon, France. 4Department of Linguistics and School of Languages, Literatures & Cultures, University of Maryland, College Park, USA. 5Department of Spanish, Linguistics, and Theory of Literature (Linguistics). Faculty of Philology. University of Seville, Seville, Spain * Corresponding author: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/434209; this version posted October 3, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. ABSTRACT Interferon-gamma (IFNa prototypical T lymphocyte-derived pro-inflammatory cytokine, was recently shown to shape social behavior and neuronal connectivity in rodents. STAT1 (Signal Transducer And Activator Of Transcription 1) is a transcription factor (TF) crucially involved in the IFN pathway. -

Mouse Ifi27l2a Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ifi27l2a Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Ifi27l2a conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ifi27l2a gene (NCBI Reference Sequence: NM_029803 ; Ensembl: ENSMUSG00000079017 ) is located on Mouse chromosome 12. 4 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 4 (Transcript: ENSMUST00000055071). Exon 3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Ifi27l2a gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP24-62E20 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit normal susceptibility to infection with H1N1 or H7N7 influenza A virus, although sloughing of dead cells into bronchiolar lumen persists somewhat longer after infection than in infected controls. Exon 3 is not frameshift exon, and covers 60.0% of the coding region. The size of intron 2 for 5'-loxP site insertion: 488 bp, and the size of intron 3 for 3'-loxP site insertion: 448 bp. The size of effective cKO region: ~630 bp. The cKO region does not have any other known gene. Page 1 of 7 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 3 4 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Homology arm Exon of mouse Ifi27l2a cKO region loxP site Page 2 of 7 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Supplementary Data
Supplemental figures Supplemental figure 1: Tumor sample selection. A total of 98 thymic tumor specimens were stored in Memorial Sloan-Kettering Cancer Center tumor banks during the study period. 64 cases corresponded to previously untreated tumors, which were resected upfront after diagnosis. Adjuvant treatment was delivered in 7 patients (radiotherapy in 4 cases, cyclophosphamide- doxorubicin-vincristine (CAV) chemotherapy in 3 cases). 34 tumors were resected after induction treatment, consisting of chemotherapy in 16 patients (cyclophosphamide-doxorubicin- cisplatin (CAP) in 11 cases, cisplatin-etoposide (PE) in 3 cases, cisplatin-etoposide-ifosfamide (VIP) in 1 case, and cisplatin-docetaxel in 1 case), in radiotherapy (45 Gy) in 1 patient, and in sequential chemoradiation (CAP followed by a 45 Gy-radiotherapy) in 1 patient. Among these 34 patients, 6 received adjuvant radiotherapy. 1 Supplemental Figure 2: Amino acid alignments of KIT H697 in the human protein and related orthologs, using (A) the Homologene database (exons 14 and 15), and (B) the UCSC Genome Browser database (exon 14). Residue H697 is highlighted with red boxes. Both alignments indicate that residue H697 is highly conserved. 2 Supplemental Figure 3: Direct comparison of the genomic profiles of thymic squamous cell carcinomas (n=7) and lung primary squamous cell carcinomas (n=6). (A) Unsupervised clustering analysis. Gains are indicated in red, and losses in green, by genomic position along the 22 chromosomes. (B) Genomic profiles and recurrent copy number alterations in thymic carcinomas and lung squamous cell carcinomas. Gains are indicated in red, and losses in blue. 3 Supplemental Methods Mutational profiling The exonic regions of interest (NCBI Human Genome Build 36.1) were broken into amplicons of 500 bp or less, and specific primers were designed using Primer 3 (on the World Wide Web for general users and for biologist programmers (see Supplemental Table 2) [1]. -

Comprehensive Epigenomic Profiling of Human Alveolar Epithelial
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.14.439786; this version posted April 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. FULL TITLE: Comprehensive epigenomic profiling of human alveolar epithelial differentiation identifies key epigenetic states and transcription factor co-regulatory networks for maintenance of distal lung identity SHORT TITLE: Epigenomic profiling of human alveolar epithelial differentiation Beiyun Zhou1,2,3, Theresa Ryan Stueve3,4,5, Evan A. Mihalakakos3,4,5, Lin Miao3,4,5, Daniel J Mullen3,4,5, Yinchong Wang3,4,5, Yixin Liu1, Jiao Luo1, Evelyn Tran3,4,5, Kim D. Siegmund6, Sean K. Lynch7, Amy L. Ryan1,2,8, Ite A. Offringa3,4,5,#, Zea Borok1,2,3,5,9,#, Crystal N. Marconett3,4,5* 1Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, 2Hastings Center for Pulmonary Research, 3Norris Comprehensive Cancer Center, Departments of 4Surgery, 5Biochemistry and Molecular Medicine, and 6Preventive Medicine, Keck School of Medicine, University of Southern California, USA 90089 7Department of Engineering, Test Manufacturing Group, MAXIM Integrated Products, Sunnyvale, California, USA 95134 8Department of Stem Cell Biology and Regenerative Medicine, University of Southern California, Los Angeles, CA USA 90089 9Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of California, San Diego, La Jolla, CA USA 92093. #These authors contributed equally * Corresponding/Senior author contact information: Crystal Marconett, Ph.D., Norris Comprehensive Cancer Center, Keck School of Medicine, University of Southern California, NTT 6418A M/C 9176, Los Angeles CA 90089-9520, USA. -

AP-1 Imprints a Reversible Transcriptional Program of Senescent Cells
bioRxiv preprint doi: https://doi.org/10.1101/633594; this version posted May 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. AP-1 Imprints a Reversible Transcriptional Program of Senescent Cells Ricardo Iván Martínez-Zamudio1,5,8, Pierre-François Roux1,5, José Américo N L F de Freitas1,4, Lucas Robinson1,4, Gregory Doré1, Bin Sun6,7, Jesús Gil6,7, Utz Herbig8 and Oliver Bischof1,2,3,9* 1 Institut Pasteur, Laboratory of Nuclear Organization and Oncogenesis, Department of Cell Biology and Infection, 75015 Paris, France 2 INSERM, U993, 75015 Paris, France 3 Equipe Labellisée Fondation ARC pour la recherche sur le cancer, Paris, France 4 Université de Paris, Sorbonne Paris Cité, Paris, France 5 These authors contributed equally to this work 6 MRC London Institute of Medical Sciences (LMS), Du Cane Road, London, W12 0NN, UK. 7 Institute of Clinical Sciences (ICS), Faculty of Medicine, Imperial College London, Du Cane Road, London W12 0NN, UK. 8 Department of Microbiology, Biochemistry & Molecular Genetics, Rutgers Biomedical & Health Sciences, Rutgers University, 205 South Orange Avenue, Newark, NJ 07103, USA 9 Lead contact *Correspondence: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/633594; this version posted May 9, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. SUMMARY Senescent cells play important physiological- and pathophysiological roles in tumor suppression, tissue regeneration, and aging.