Supporting Material 1,2,4,3-Triazaborole-Based Neutral
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Connecting Bangladesh: Economic Corridor Network
Connecting Bangladesh: Economic Corridor Network Economic corridors are anchored on transport corridors, and international experience suggests that the higher the level of connectivity within and across countries, the higher the level of economic growth. In this paper, a new set of corridors is being proposed for Bangladesh—a nine-corridor comprehensive integrated multimodal economic corridor network resembling the London Tube map. This paper presents the initial results of the research undertaken as an early step of that development effort. It recommends an integrated approach to developing economic corridors in Bangladesh that would provide a strong economic foundation for the construction of world-class infrastructure that, in turn, could support the growth of local enterprises and attract foreign investment. About the Asian Development Bank COnnecTING BANGLADESH: ADB’s vision is an Asia and Pacific region free of poverty. Its mission is to help its developing member countries reduce poverty and improve the quality of life of their people. Despite the region’s many successes, it remains home to a large share of the world’s poor. ADB is committed to reducing poverty through inclusive economic growth, environmentally sustainable growth, and regional integration. ECONOMIC CORRIDOR Based in Manila, ADB is owned by 67 members, including 48 from the region. Its main instruments for helping its developing member countries are policy dialogue, loans, equity investments, guarantees, grants, NETWORK and technical assistance. Mohuiddin Alamgir -

Connecting South Asia and Southeast Asia: a Bangladesh Country Study
ADBI Working Paper Series Connecting South Asia and Southeast Asia: A Bangladesh Country Study Mustafizur Rahman, Khondaker Golam Moazzem, Mehruna Islam Chowdhury, and Farzana Sehrin No. 500 September 2014 Asian Development Bank Institute Mustafizur Rahman is executive director, Centre for Policy Dialogue (CPD), Bangladesh. Khondaker Golam Moazzem is an additional research director, CPD. Mehruna Islam Chowdhury is a senior research associate, CPD. Farzana Sehrin is a research associate, CPD. The authors would like to thank the paper reviewers and participants at the meeting organized by ADB in Manila, 6–7 November 2013. They also would like to express their sincere appreciation for the support provided by the ADBI staff who helped in preparing this manuscript for publication. The views expressed in this paper are the views of the authors and do not necessarily reflect the views or policies of ADBI, ADB, its Board of Directors, or the governments they represent. ADBI does not guarantee the accuracy of the data included in this paper and accepts no responsibility for any consequences of their use. Terminology used may not necessarily be consistent with ADB official terms. Working papers are subject to formal revision and correction before they are finalized and considered published. The Working Paper series is a continuation of the formerly named Discussion Paper series; the numbering of the papers continued without interruption or change. ADBI’s working papers reflect initial ideas on a topic and are posted online for discussion. ADBI encourages readers to post their comments on the main page for each working paper (given in the citation below). -
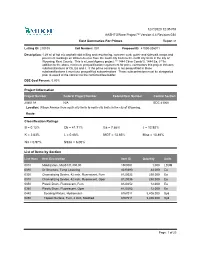
Classification Ratings Project Information B = 0.12% Cb = 41.71
12/7/2020 12:35 PM AASHTOWare Project™ Version 4.4 Revision 034 Cost Summaries For Primes Report v1 Letting ID: 210108 Call Number: 001 Proposal ID: 41000-206011 Description: 1.29 mi of hot mix asphalt cold milling and resurfacing, concrete curb, gutter and sidewalk ramps and pavement markings on Wilson Avenue from the south city limits to the north city limits in the city of Wyoming, Kent County. This is a Local Agency project. ** 1444 Cb or Comb/Jt. 1444 Ea, J **In addition to the above minimum prequalification requirement for prime contractors this project includes subclassifications of Cb, Ea and J. If the prime contractor is not prequalified in those subclassifications it must use prequalified subcontractors. Those subcontractors must be designated prior to award of the contract to the confirmed low bidder. DBE Goal Percent: 0.00% Project Information Project Number Federal Project Number Federal Item Number Control Section 206011A N/A EDC 41000 Location: Wilson Avenue from south city limits to north city limits in the city of Wyoming. Route: Classification Ratings B = 0.12% Cb = 41.71% Ea = 7.86% J = 12.92% K = 2.83% L = 0.48% MOT = 13.65% Misc = 13.46% N3 = 0.97% N93A = 6.00% List of Items by Section Line Num Item Description Item ID Quantity Units 0010 Mobilization, Max$131,300.00 1500001 1.000 LSUM 0090 Dr Structure, Temp Lowering 4030390 44.000 Ea 0300 Channelizing Device, 42 inch, Fluorescent, Furn 8120035 250.000 Ea 0310 Channelizing Device, 42 inch, Fluorescent, Oper 8120036 250.000 Ea 0350 Plastic Drum, Fluorescent, Furn -
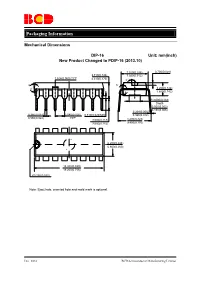
Mechanical-Dimensions-BCD.Pdf
Packaging Information Mechanical Dimensions DIP-16 Unit: mm(inch) New Product Changed to PDIP-16 (2013.10) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060) TYP 4.310(0.170) 5° 6° 6° 4° 3.200(0.126) 4° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014) 0.560(0.022) TYP 3.000(0.118) 8.200(0.323) 3.600(0.142) 9.400(0.370) 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optional. Dec. 2014 BCD Semiconductor Manufacturing Limited Packaging Information Mechanical Dimensions DIP-16 (Special for AZ4052) Unit: mm(inch) 7.320(0.288) 0.700(0.028) 3.710(0.146) 7.920(0.312) 1.524(0.060)T YP 4.310(0.170) 5 ° 6 ° 6 ° 4 ° 3.200(0.126) 4 ° 3.600(0.142) Φ3.000(0.118) Depth 0.050(0.002) 0.150(0.006) 0.204(0.008) 0.360(0.014) 2.540(0.100) 0.510(0.020)MIN 0.360(0.014 ) 0.560(0.022) TYP 3.000(0.118) 8.500(0.335) 3.600(0.142) TYP 6.200(0.244) 6.600(0.260) 18.800(0.740) 19.200(0.756) R0.750(0.030) Note: Eject hole, oriented hole and mold mark is optiona.l Dec. -

The Completeness and Vulnerability of Road Network in Bangladesh
M. A. Ali, S. M. Seraj and S. Ahmad (eds): ISBN 984-823-002-5 The Completeness and Vulnerability of Road Network in Bangladesh A. S. M. Abdul Quium and S. A. M. Aminul Hoque Department of Urban and Regional Planning Bangladesh University of Engineering and Technology, Dhaka-1000, Bangladesh Abstract Road transportation has emerged as the most popular mode of transportation in Bangladesh. Despite being the most popular mode, it suffers heavily from network failures due to natural as well as man-made disruptions. This paper presents the major findings of a study relating to the completeness of the road network with respect to the pattern of traffic flow and its vulnerability with reference to the disruptions caused by the flood of 1998. The study considered development of road network by regions on the basis of degree of network completeness by connectivity indices namely, γ and α indices that apply the Graph Theory to measure the geometric pattern of a network. The analysis revealed that the road network development in Bangladesh at the national and regional levels is still in its early stage. The existing network has a very few circuits for movement at all the levels and, therefore, susceptible to high risk of disruption. In terms of circuitry, the network has a bare marginal grid configuration at the divisional and national levels. The present overwhelming dependence on road transportation system will continue in the future. In this context, completeness of the whole road network should be considered in all future road planning exercises. Assessment of vulnerability of links around Dhaka deserves special consideration as any link failure of the national highways around Dhaka affects almost the whole system. -

Land Resource Appraisal of Bangladesh for Agricultural
BGD/81/035 Technical Report 3 Volume II LAND RESOURCES APPRAISAL OF BANGLADESH FOR AGRICULTURAL DEVELOPMENT REPORT 3 LAND RESOURCES DATA BASE VOLUME II SOIL, LANDFORM AND HYDROLOGICAL DATA BASE A /UNITED NATIONS DEVELOPMENT PROGRAMME FAo FOOD AND AGRICULTURE ORGANIZATION vJ OF THE UNITED NATIONS BGD/81/035 Technical Report 3 Volume II LAND RESOURCES APPRAISAL OF BANGLADESH FOR AGRICULTURALDEVELOPMENT REPORT 3 LAND RESOURCES DATA BASE VOLUME II SOIL, LANDFORM AND HYDROLOGICAL DATA BASE Report prepared for the Government of the People's Republic of Bangladesh by the Food and Agriculture Organization of the United Nations acting as executing agency for the United Nations Development Programme based on the work of H. Brammer Agricultural Development Adviser J. Antoine Data Base Management Expert and A.H. Kassam and H.T. van Velthuizen Land Resources and Agricultural Consultants UNITED NATIONS DEVELOPMENT PROGRAMME FOOD AND AGRICULTURE ORGANIZATION OF THE UNITED NATIONS Rome, 1988 The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and AgricultureOrganization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. All rights reserved. No part of this publication may be reproduced, stored ina retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopyingor otherwise, without the prior perrnission of (he copyright owner. Applications for such permission,with a statement of the purpose and extent of the reproduction, should be addressedto the Director, Publications Division, Food and Agriculture Organization of the United Nations, Viadelle Terme di Caracarla, 00100 Home, Italy. -

Bangladesh Agron
Bangladesh Agron. J. 2015, 18(1): 89-98 GROWTH, YIELD AND QUALITY OF WHEAT VARITIES AS AFFECTED BY DIFFERENT LEVELS OF NITROGEN H. Mondal1, S. Mazumder2, S. K. Roy3, T. A. Mujahidi4 and S. K. Paul5 1Department of Agronomy, Sher-e-Bangla Agricultural University, Dhaka-1207, Bangladesh 2Sher-e-Bangla Nagar Adorsha Mohila College, Dhaka-1207, Bangladesh, 3Department of Genetics and Plant Breeding, Sher-e-Bangla Agricultural University, Dhaka-1207, Bangladesh 4Plant Breeding Division, Bangladesh Agricultural Research Institute, Joydebpur, Gazipur-1701, Bangladesh 5Agronomy Division, Bangladesh Agricultural Research Institute, Joydebpur, Gazipur-1701, Bangladesh. E-mail: [email protected] Key Words: Wheat variety, Nitrogen, Protein, Grain quality Abstract A field experiment was conducted at the experimental field of Sher-e-Bangla Agricultural University, Dhaka, Bangladesh during November 2012 to March 2013 to evaluate the response of three (3) wheat varieties viz., BARI Gom23, BARI Gom24 and BARI Gom25 under four levels of nitrogen fertilizer i.e, 75, 100, 125 and 150 kg N ha-1. The experiment was laid out in Randomized Complete Block Design (RCBD) with three replications. Results showed that plant height, number of leaves plant-1, leaf length and dry matter content were significantly affected due to varieties and/or nitrogen levels. Grains ear-1, number of fertile grains plant-1, 1000-grain weight, grain yield and harvest index were also significantly influenced by varieties and/or nitrogen levels. The value of all parameters studied in this experiment increased with increasing nitrogen levels up to 125 kg N ha-1 and thereafter decreased with fertilizer increasing level. Combination results showed that BARI Gom-24 with application of 125 kg N ha-1 gave the maximum grain yield (4.71 t ha-1), harvest index (49.37 %) and protein content (10.88%). -

ASEAN Connectivity, Project Information Sheet
ASEAN Connectivity Project Information Sheets one vision one identity one community The Association of Southeast Asian Nations (ASEAN) was established on 8 August 1967. The Member States of the Association are Brunei Darussalam, Cambodia, Indonesia, Lao PDR, Malaysia, Myanmar, Philippines, Singapore, Thailand and Viet Nam. The ASEAN Secretariat is based in Jakarta, Indonesia. The ASEAN Secretariat Public Outreach and Civil Society Division 70A Jalan Sisingamangaraja Jakarta 12110, Indonesia Phone : (62 21) 724-3372, 726-2991 Fax : (62 21) 739-8234, 724-3504 E-mail : [email protected] General information on ASEAN appears online at the ASEAN Website: www.asean.org Catalogue-in-Publication Data ASEAN Connectivity Jakarta: ASEAN Secretariat, August 2012 The text of this publication may be freely quoted or reprinted with proper acknowledgement. Copyright Association of Southeast Asian Nations (ASEAN) 2012 All rights reserved PHYSICAL CONNECTIVITY PrOjECT INfOrmATION SHEET mPAC PP/A1/01 CONSTRUCTION OF THE ASEAN HIGHWAY NETWORK (AHN) MISSING LINKS AND UPGRADE OF TRANSIT TRANSPORT ROUTES IN LAO PDR AND MYANMAR PrOjECT DESCrIPTION PrOjECT STATuS ASEAN HIGHWAY Given the growing regional integration and cooperation Seeking technical in ASEAN, the development of high quality transport assistance and funding NETWORK infrastructure is a crucial element to building a competitive ASEAN Community with equitable economic TArgET COmPLETION development. However, ASEAN with total land area DATE of around 4.4 million sq. km faces challenges with poor December 2015 quality of roads and incomplete road networks. The ASEAN Highway Network (AHN) project is a flagship ImPLEmENTINg bODIES infrastructure project that seeks to bring connectivity Ministry of Public across borders and confer many benefits, such as Works and Transport improved competitiveness of regional production of Lao PDR, Ministry networks, better trade and investment flows, and of Construction of reductions in development gaps. -

X-Ray Single Crystal Structure, Tautomerism Aspect, DFT, NBO
crystals Article X-ray Single Crystal Structure, Tautomerism Aspect, DFT, NBO, and Hirshfeld Surface Analysis of a New Schiff Bases Based on 4-Amino-5-Indol-2-yl-1,2,4-Triazole-3-Thione Hybrid Ahmed T. A. Boraei 1,*, Saied M. Soliman 2 , Matti Haukka 3 , El Sayed H. El Tamany 1 , Abdullah Mohammed Al-Majid 4 and Assem Barakat 4,* 1 Chemistry Department, Faculty of Science, Suez Canal University, Ismailia 41522, Egypt; [email protected] 2 Department of Chemistry, Faculty of Science, Alexandria University, P.O. Box 426, Ibrahimia, Alexandria 21321, Egypt; [email protected] or [email protected] 3 Department of Chemistry, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland; matti.o.haukka@jyu.fi 4 Department of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia; [email protected] * Correspondence: [email protected] (A.T.A.B.); [email protected] (A.B.); Tel.: +966-11467-5901 (A.B.); Fax: +966-11467-5992 (A.B.) Abstract: Four different new Schiff basses tethered indolyl-triazole-3-thione hybrid were designed Citation: Boraei, A.T.A.; Soliman, and synthesized. X-ray single crystal structure, tautomerism, DFT, NBO and Hirshfeld analysis were S.M.; Haukka, M.; El Tamany, E.S.H.; explored. X-ray crystallographic investigations with the aid of Hirshfeld calculations were used Al-Majid, A.M.; Barakat, A. X-ray to analyze the molecular packing of the studied systems. The H···H, H···C, S···H, Br···C, O···H, Single Crystal Structure, C···C and N···H interactions are the most important in the molecular packing of 3. -

Computational Methods
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 + Structural Evolution of LiNn (n=2, 4, 6, 8, and 10) Clusters: Mass Spectrometry and Theoretical Calculations Zhongxue Ge,*,a,b Kewei Ding,a,b Yisu Li,c Hongguang Xu,d Zhaoqiang Chen,c Yiding Ma,b Taoqi Li,b Weiliang Zhu,*,c and Weijun Zheng*,d a State Key Laboratory of Fluorine & Nitrogen Chemicals, Xi’an 710065, China b Xi’an Modern Chemistry Research Institute, Xi’an 710065, China c Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China d Beijing National Laboratory for Molecular Sciences (BNLMS), State Key Laboratory of Molecular Reaction Dynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China 1 Fig.S1 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF target with N2 as carrier gas Fig.S2 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF:ZrN=4:1 mixture target with N2 as carrier gas Fig.S3 Typical mass spectrum of Li-N clusters generated by laser ablation of a 2 LiF:AlN=2:1 mixture target with N2 as carrier gas Fig.S4 Typical mass spectrum of Li-N clusters generated by laser ablation of a LiF:BN=2:1 mixture target with N2 as carrier gas 3 + Table S1 NBO data for LiN2 (LP: lone pair; BD: bonding; BD*: antibonding;E2: stabilization energy) NPA(Natural Population Analysis) Atom Natural Electron Configuration Natural Charge Li 2s0.012p0.01 0.976 N2 2s1.572p3.573s0.033p0.01 -0.193 -

On the Existence of Partitioned Incomplete Latin Squares with Five Parts
Marshall University Marshall Digital Scholar Mathematics Faculty Research Mathematics 2019 On the Existence of Partitioned Incomplete Latin Squares with Five Parts Jaromy Kuhl Donald McGinn Michael W. Schroeder Follow this and additional works at: https://mds.marshall.edu/mathematics_faculty Part of the Discrete Mathematics and Combinatorics Commons AUSTRALASIAN JOURNAL OF COMBINATORICS Volume 74(1) (2019), Pages 46–60 On the existence of partitioned incomplete Latin squares with five parts Jaromy Kuhl Donald McGinn Department of Mathematics and Statistics University of West Florida Pensacola, FL 32514 U.S.A. [email protected] [email protected] Michael William Schroeder Department of Mathematics Marshall University Huntington, WV 25755 U.S.A. [email protected] Abstract Let a, b, c, d,ande be positive integers. In 1982 Heinrich showed the existence of a partitioned incomplete Latin square (PILS) of type (a, b, c) and (a, b, c, d) if and only if a = b = c and 2a ≥ d. For PILS of type (a, b, c, d, e) with a ≤ b ≤ c ≤ d ≤ e, it is necessary that a + b + c ≥ e, but not sufficient. In this paper we prove an additional necessary condition and classify the existence of PILS of type (a, b, c, d, a + b + c)andPILS with three equal parts. Lastly, we show the existence of a family of PILS in which the parts are nearly the same size. 1 Introduction Let a, n ∈ Z+ and S be a symbol set of order n.Let[n]={1,...,n}, a+S = {a+s | s ∈ S},andaS be the multiset in which each element of S occurs a times. -

Antigenic and Genetic Characteristics of Zoonotic Influenza Viruses and Development of Candidate Vaccine Viruses for Pandemic Preparedness
4 Antigenic and genetic characteristics of zoonotic influenza viruses and development of candidate vaccine viruses for pandemic preparedness February 2019 The development of influenza candidate vaccine viruses (CVVs), coordinated by WHO, remains an essential component of the overall global strategy for pandemic preparedness. Selection and development of CVVs are the first steps towards timely vaccine production and do not imply a recommendation for initiating manufacture. National authorities may consider the use of one or more of these CVVs for pilot lot vaccine production, clinical trials and other pandemic preparedness purposes based on their assessment of public health risk and need. Zoonotic influenza viruses continue to be identified and evolve both genetically and antigenically, leading to the need for additional CVVs for pandemic preparedness purposes. Changes in the genetic and antigenic characteristics of these viruses relative to existing CVVs, and their potential risks to public health justify the need to select and develop new CVVs. This document summarizes the genetic and antigenic characteristics of recent zoonotic influenza viruses and related viruses circulating in animals1 that are relevant to CVV updates. Institutions interested in receiving these CVVs should contact WHO at [email protected] or the institutions listed in announcements published on the WHO website2. Influenza A(H5) Since their emergence in 1997, highly pathogenic avian influenza (HPAI) A(H5) viruses of the A/goose/Guangdong/1/96 haemagglutinin (HA) lineage have become enzootic in some countries, have infected wild birds and continue to cause outbreaks in poultry and sporadic human infections. These viruses have diversified genetically and antigenically, including the emergence of viruses with replacement of the N1 gene segment by N2, N3, N5, N6, N8 or N9 gene segments, leading to the need for multiple CVVs.