2018 Chen Lingyan 1448129
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MOUSE INTERLEUKIN-28B/INTERFERON-LAMBDA 3, CARRIER FREE Product Number: 12821-1 Lot Number: 5559 Size: 25 Μg
MOUSE INTERLEUKIN-28B/INTERFERON-LAMBDA 3, CARRIER FREE Product Number: 12821-1 Lot Number: 5559 Size: 25 µg Description: Mouse Interleukin-28B/Interferon-lambda 3, Carrier Free Source: A DNA sequence encoding the mature mouse IL-28B/IFN-λ3 (Asp 20 - Val 193) (Kotenko, S.V. et al., 2003, Nat. Immunol. 4(1):69 - 77) was expressed in E. coli. Form: Lyophilized Buffer: Phosphate-buffered saline (PBS) Reconstitution: It is recommended that sterile PBS be added to the vial to prepare a stock solution of no less than 100 μg/mL. Endotoxin: < 1 EU/µg Molecular Weight: The 175 amino acid residue methionyl form of recombinant mouse IL-28B has a predicted molecular mass of approximately 19.7 kDa. Purity: > 95% Synonyms: Mu IL-28B; Mu IFN-λ3 Accession #: NP_796370 Assays Used to Measure Bioactivity: Human HepG2 cells infected with encephalomyocarditis virus (Sheppard, P. et al., 2003, Nature Immunol. 4:63). The ED50 for this effect is typically 7.5 - 37.5 ng/mL. Shipping Conditions: Wet Ice Physical State of Product During Shipping: Lyophilized Special Conditions/Comments: After receipt, this product should be kept at -70˚C or below for retention of full activity. Upon reconstitution, this cytokine can be stored under sterile conditions at 2˚C to 8˚C for one month or at -20˚C to -70˚C in a manual defrost freezer for three months without detection loss of activity. Avoid repeated freeze-thaw cycles. For more information on protein handling, visit the PBL website at www.interferonsource.com . Product Information: Human IL-28A, IL-28B, and IL-29, also named interferon-λ2 (IFN-λ2), IFN-λ3, and IFN-λ1, respectively, are newly identified class II cytokine receptor ligands that are distantly related to members of the IL-10 family (11- 13% aa sequence identity) and type I IFN family (15 - 19% aa sequence identity).1 – 3 The genes encoding these three cytokines are localized to chromosome 19 and each is composed of multiple exons. -

Molecular Characterization of Tea Catechin Treated Human Prostate Cancer Cell Lines Yewseok Suh
Florida State University Libraries Electronic Theses, Treatises and Dissertations The Graduate School 2006 Molecular Characterization of Tea Catechin Treated Human Prostate Cancer Cell Lines Yewseok Suh Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected] THE FLORIDA STATE UNIVERSITY COLLEGE OF ARTS AND SCIENCES MOLECULAR CHARACTERIZATION OF TEA CATECHIN TREATED HUMAN PROSTATE CANCER CELL LINES By YEWSEOK SUH A Dissertation submitted to the Department of Chemistry and Biochemistry in partial fulfillment of the requirements for the degree of Doctor of Philosophy Degree Awarded: Summer Semester, 2006 The members of the Committee approve the dissertation of Yewseok Suh defended on May 12, 2006. Qing-Xiang Amy Sang Professor Directing Dissertation Thomas C.S. Keller III Outside Committee Member Joseph B. Schlenoff Committee Member Hong Li Committee Member Approved: Naresh Dalal, Chair, Department of Chemistry and Biochemistry Joseph Travis, Dean, College of Arts and Sciences The Office of Graduate Studies has verified and approved the above named committee members. ii This dissertation is dedicated to my parents for their endless love and encouragement, to my lovely wife Inok Park for her support, and to our charming daughter, Tae-won. iii ACKNOWLEDGEMENTS I would like to express my gratitude to my major professor for her support throughout the research. Also thanks to all my committee members, Dr. Thomas C. S. Keller III, Dr. Hong Li, and Dr. Joseph B. Schlenoff, for their support, advice, and guidance. Special thanks to our lab members especially, Ziad Sahab and Robert G. Newcomer and former lab member Douglas R. -
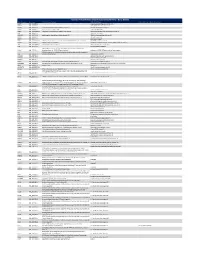
Ncounter® Autoimmune Discovery Consortium Panel
nCounter® Autoimmune Discovery Consortium Panel - Gene Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information AAMP NM_001087.3 angio associated migratory cell protein ABHD6 NM_020676.5 abhydrolase domain containing 6 ACKR2 NM_001296.3 CMKBR9,CCBP2;chemokine binding protein 2 atypical chemokine receptor 2 ACOXL NM_018308.1 acyl-Coenzyme A oxidase-like acyl-CoA oxidase like ACSL6 NM_001009185.1 FACL6;fatty-acid-Coenzyme A ligase, long-chain 6 acyl-CoA synthetase long chain family member 6 ADA NM_000022.2 adenosine deaminase ADAM30 NM_021794.2 a disintegrin and metalloproteinase domain 30 ADAM metallopeptidase domain 30 ADCY3 NM_004036.3 adenylate cyclase 3 ADCY7 NM_001114.4 adenylate cyclase 7 AFF3 NM_001025108.1 LAF4;lymphoid nuclear protein related to AF4,AF4/FMR2 family, member 3 AF4/FMR2 family member 3 AGAP2 NM_014770.3 CENTG1;centaurin, gamma 1 ArfGAP with GTPase domain, ankyrin repeat and PH domain 2 AHI1 NM_001134830.1 Abelson helper integration site Abelson helper integration site 1 AHR NM_001621.3 aryl hydrocarbon receptor AHSA2;AHA1, activator of heat shock 90kDa protein ATPase homolog 2 AHSA2 NM_152392.1 (yeast),activator of HSP90 ATPase homolog 2 activator of HSP90 ATPase homolog 2, pseudogene APECED;autoimmune regulator (autoimmune polyendocrinopathy candidiasis AIRE NM_000383.2 ectodermal dystrophy) autoimmune regulator AMIGO3 NM_198722.2 adhesion molecule with Ig like domain 3 ANKRD55 NM_024669.2 ankyrin repeat domain 55 ANTXR2 NM_058172.5 anthrax toxin receptor -

Review of Lambda Interferons in Hepatitis B Virus Infection: Outcomes and Therapeutic Strategies
viruses Review Review of Lambda Interferons in Hepatitis B Virus Infection: Outcomes and Therapeutic Strategies Laura A. Novotny 1 , John Grayson Evans 1, Lishan Su 2, Haitao Guo 3 and Eric G. Meissner 1,4,* 1 Division of Infectious Diseases, Medical University of South Carolina, Charleston, SC 29525, USA; [email protected] (L.A.N.); [email protected] (J.G.E.) 2 Division of Virology, Pathogenesis, and Cancer, Institute of Human Virology, Departments of Pharmacology, Microbiology, and Immunology, University of Maryland School of Medicine, Baltimore, MD 21201, USA; [email protected] 3 Department of Microbiology and Molecular Genetics, Cancer Virology Program, UPMC Hillman Cancer Center, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA; [email protected] 4 Department of Microbiology and Immunology, Medical University of South Carolina, Charleston, SC 29425, USA * Correspondence: [email protected]; Tel.: +1-843-792-4541 Abstract: Hepatitis B virus (HBV) chronically infects over 250 million people worldwide and causes nearly 1 million deaths per year due to cirrhosis and liver cancer. Approved treatments for chronic infection include injectable type-I interferons and nucleos(t)ide reverse transcriptase inhibitors. A small minority of patients achieve seroclearance after treatment with type-I interferons, defined as sustained absence of detectable HBV DNA and surface antigen (HBsAg) antigenemia. However, type-I interferons cause significant side effects, are costly, must be administered for months, and most patients have viral rebound or non-response. Nucleos(t)ide reverse transcriptase inhibitors reduce HBV viral load and improve liver-related outcomes, but do not lower HBsAg levels or impart Citation: Novotny, L.A.; Evans, J.G.; seroclearance. -

Cytokine-Targeted Therapeutics for KSHV-Associated Disease
viruses Review Cytokine-Targeted Therapeutics for KSHV-Associated Disease Nedaa Alomari and Jennifer Totonchy * Department of Biomedical and Pharmaceutical Sciences, Chapman University School of Pharmacy, Irvine, CA 92618, USA; [email protected] * Correspondence: [email protected] Received: 1 September 2020; Accepted: 25 September 2020; Published: 28 September 2020 Abstract: Kaposi’s sarcoma-associated herpesvirus (KSHV) also known as human herpesvirus 8 (HHV-8), is linked to several human malignancies including Kaposi sarcoma (KS), primary effusion lymphoma (PEL), multicentric Castleman’s disease (MCD) and recently KSHV inflammatory cytokine syndrome (KICS). As with other diseases that have a significant inflammatory component, current therapy for KSHV-associated disease is associated with significant off-target effects. However, recent advances in our understanding of the pathogenesis of KSHV have produced new insight into the use of cytokines as potential therapeutic targets. Better understanding of the role of cytokines during KSHV infection and tumorigenesis may lead to new preventive or therapeutic strategies to limit KSHV spread and improve clinical outcomes. The cytokines that appear to be promising candidates as KSHV antiviral therapies include interleukins 6, 10, and 12 as well as interferons and tumor necrosis factor-family cytokines. This review explores our current understanding of the roles that cytokines play in promoting KSHV infection and tumorigenesis, and summarizes the current use of cytokines as therapeutic targets in KSHV-associated diseases. Keywords: KSHV; pathogenesis; cytokine signaling; cytokine therapeutics; cytokine targeted therapy; immunomodulatory therapeutics 1. Introduction In 1994, Kaposi’s sarcoma-associated herpesvirus (KSHV) was first identified in a Kaposi’s sarcoma (KS) lesion by Chang and Moore [1]. -

COVID-19: a Drug Repurposing and Biomarker Identification by Using Comprehensive Gene-Disease Associations Through Protein-Protein Interaction Network Analysis
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 30 March 2020 doi:10.20944/preprints202003.0440.v1 COVID-19: A drug repurposing and biomarker identification by using comprehensive gene-disease associations through protein-protein interaction network analysis Suresh Kumar* Department of Diagnostic and Allied Health Science, Faculty of Health & Life Sciences, Management & Science University, 40100 Shah Alam, Selangor, Malaysia *Corresponding author Suresh Kumar, PhD Department of Diagnostic and Allied Health Science, Faculty of Health & Life Sciences Management & Science University, 40100 Shah Alam, Selangor, Malaysia Email: [email protected] Abstract COVID-19 (2019-nCoV) is a pandemic disease with an estimated mortality rate of 3.4% (estimated by the WHO as of March 3, 2020). Until now there is no antiviral drug and vaccine for COVID-19. The current overwhelming situation by COVID-19 patients in hospitals is likely to increase in the next few months. About 15 percent of patients with serious disease in COVID-19 require immediate health services. Rather than waiting for new anti-viral drugs or vaccines that take a few months to years to develop and test, several researchers and public health agencies are attempting to repurpose medicines that are already approved for another similar disease and have proved to be fairly effective. This study aims to identify FDA approved drugs that can be used for drug repurposing and identify biomarkers among high- risk and asymptomatic groups. In this study gene-disease association related to COVID-19 reported mild, severe symptoms and clinical outcomes were determined. The high-risk group was studied related to SARS-CoV-2 viral entry and life cycle by using Disgenet and compared with curated COVID-19 gene data sets from the CTD database. -

R J M E EVIEW Romanian Journal of R Morphology & Embryology
Rom J Morphol Embryol 2020, 61(1):33–43 doi: 10.47162/RJME.61.1.04 R J M E EVIEW Romanian Journal of R Morphology & Embryology http://www.rjme.ro/ Host immune response in chronic hepatitis C infection: involvement of cytokines and inflammasomes MIHAIL VIRGIL BOLDEANU1,2), ISABELA SILOŞI1), ANDREEA LILI BĂRBULESCU3), RALUCA ELENA SANDU4), CRISTIANA GEORMĂNEANU5), VLAD PĂDUREANU6), MIRCEA VASILE POPESCU-DRIGĂ7), IOAN SABIN POENARIU7), CRISTIAN ADRIAN SILOŞI8), ANCA MARILENA UNGUREANU9), ANDA LORENA DIJMĂRESCU10), LIDIA BOLDEANU9) 1)Department of Immunology, University of Medicine and Pharmacy of Craiova, Romania 2)Medico Science SRL – Stem Cell Bank Unit, Craiova, Romania 3)Department of Pharmacology, University of Medicine and Pharmacy of Craiova, Romania 4)Department of Biochemistry, University of Medicine and Pharmacy of Craiova, Romania 5)Department of Emergency Medicine and First Aid, University of Medicine and Pharmacy of Craiova, Romania 6)Department of Internal Medicine, University of Medicine and Pharmacy of Craiova, Romania 7)PhD Student, University of Medicine and Pharmacy of Craiova, Romania 8)Department of Surgery, University of Medicine and Pharmacy of Craiova, Romania 9)Department of Microbiology, University of Medicine and Pharmacy of Craiova, Romania 10)Department of Obstetrics and Gynecology, University of Medicine and Pharmacy of Craiova, Romania Abstract Chronic liver disease is a major health issue worldwide and chronic hepatitis C (CHC) is associated with an increased risk of cirrhosis and hepatocellular carcinoma (HCC). There is evidence that the hepatitis C virus (HCV) infection is correlated with immune senescence by way of immune activation and chronic inflammation, which lead to increased metabolic and cardiovascular risk, as well as progressive liver damage. -

Control of the Physical and Antimicrobial Skin Barrier by an IL-31–IL-1 Signaling Network
The Journal of Immunology Control of the Physical and Antimicrobial Skin Barrier by an IL-31–IL-1 Signaling Network Kai H. Ha¨nel,*,†,1,2 Carolina M. Pfaff,*,†,1 Christian Cornelissen,*,†,3 Philipp M. Amann,*,4 Yvonne Marquardt,* Katharina Czaja,* Arianna Kim,‡ Bernhard Luscher,€ †,5 and Jens M. Baron*,5 Atopic dermatitis, a chronic inflammatory skin disease with increasing prevalence, is closely associated with skin barrier defects. A cy- tokine related to disease severity and inhibition of keratinocyte differentiation is IL-31. To identify its molecular targets, IL-31–dependent gene expression was determined in three-dimensional organotypic skin models. IL-31–regulated genes are involved in the formation of an intact physical skin barrier. Many of these genes were poorly induced during differentiation as a consequence of IL-31 treatment, resulting in increased penetrability to allergens and irritants. Furthermore, studies employing cell-sorted skin equivalents in SCID/NOD mice demonstrated enhanced transepidermal water loss following s.c. administration of IL-31. We identified the IL-1 cytokine network as a downstream effector of IL-31 signaling. Anakinra, an IL-1R antagonist, blocked the IL-31 effects on skin differentiation. In addition to the effects on the physical barrier, IL-31 stimulated the expression of antimicrobial peptides, thereby inhibiting bacterial growth on the three-dimensional organotypic skin models. This was evident already at low doses of IL-31, insufficient to interfere with the physical barrier. Together, these findings demonstrate that IL-31 affects keratinocyte differentiation in multiple ways and that the IL-1 cytokine network is a major downstream effector of IL-31 signaling in deregulating the physical skin barrier. -

SUPPLEMENTARY APPENDIX Exome Sequencing Reveals Heterogeneous Clonal Dynamics in Donor Cell Myeloid Neoplasms After Stem Cell Transplantation
SUPPLEMENTARY APPENDIX Exome sequencing reveals heterogeneous clonal dynamics in donor cell myeloid neoplasms after stem cell transplantation Julia Suárez-González, 1,2 Juan Carlos Triviño, 3 Guiomar Bautista, 4 José Antonio García-Marco, 4 Ángela Figuera, 5 Antonio Balas, 6 José Luis Vicario, 6 Francisco José Ortuño, 7 Raúl Teruel, 7 José María Álamo, 8 Diego Carbonell, 2,9 Cristina Andrés-Zayas, 1,2 Nieves Dorado, 2,9 Gabriela Rodríguez-Macías, 9 Mi Kwon, 2,9 José Luis Díez-Martín, 2,9,10 Carolina Martínez-Laperche 2,9* and Ismael Buño 1,2,9,11* on behalf of the Spanish Group for Hematopoietic Transplantation (GETH) 1Genomics Unit, Gregorio Marañón General University Hospital, Gregorio Marañón Health Research Institute (IiSGM), Madrid; 2Gregorio Marañón Health Research Institute (IiSGM), Madrid; 3Sistemas Genómicos, Valencia; 4Department of Hematology, Puerta de Hierro General University Hospital, Madrid; 5Department of Hematology, La Princesa University Hospital, Madrid; 6Department of Histocompatibility, Madrid Blood Centre, Madrid; 7Department of Hematology and Medical Oncology Unit, IMIB-Arrixaca, Morales Meseguer General University Hospital, Murcia; 8Centro Inmunológico de Alicante - CIALAB, Alicante; 9Department of Hematology, Gregorio Marañón General University Hospital, Madrid; 10 Department of Medicine, School of Medicine, Com - plutense University of Madrid, Madrid and 11 Department of Cell Biology, School of Medicine, Complutense University of Madrid, Madrid, Spain *CM-L and IB contributed equally as co-senior authors. Correspondence: -

Ncounter® Host Response Panel - Gene and Probe Details
nCounter® Host Response Panel - Gene and Probe Details Gene Accession Synonyms Full Name Other targets or Isoform Information ACE NM 000789.2 DCP1;angiotensin I converting enzyme (peptidyl-dipeptidase A) 1;ACE1,CD143;peptidyl-dipeptidase A angiotensin I converting enzyme ACKR2 NM 001296.5 CMKBR9,CCBP2;chemokine binding protein 2;CCR10,D6,CCR9 atypical chemokine receptor 2 ACKR3 NM 020311.1 CMKOR1,CXCR7;chemokine orphan receptor 1,chemokine (C-X-C motif) receptor 7;RDC1,GPR159 atypical chemokine receptor 3 ACKR4 NM 016557.2 CCRL1;chemokine (C-C motif) receptor-like 1;CCR11,CCBP2,VSHK1,CCX-CKR,PPR1 atypical chemokine receptor 4 ACOX1 NM 001185039.1 acyl-Coenzyme A oxidase 1, palmitoyl,acyl-CoA oxidase 1, palmitoyl;PALMCOX;palmitoyl-CoA oxidase acyl-CoA oxidase 1 FACL2;fatty-acid-Coenzyme A ligase, long-chain 2;LACS2,LACS,ACS1,LACS1,FACL1;lignoceroyl-CoA synthase,long-chain fatty-acid- ACSL1 NM_001995.4 coenzyme A ligase 1 acyl-CoA synthetase long chain family member 1 ACSL3 NM 203372.1 FACL3;fatty-acid-Coenzyme A ligase, long-chain 3;ACS3,PRO2194 acyl-CoA synthetase long chain family member 3 FACL4,MRX63,MRX68;fatty-acid-Coenzyme A ligase, long-chain 4,mental retardation, X-linked 63,mental retardation, X-linked ACSL4 NM_004458.2 68;ACS4,LACS4;lignoceroyl-CoA synthase, long-chain fatty-acid-Coenzyme A ligase 4 acyl-CoA synthetase long chain family member 4 ACVR1 NM 001105.2 ACVRLK2;activin A receptor, type I;SKR1,ALK2,ACVR1A activin A receptor type 1 ADAR NM 001025107.1 IFI4,G1P1;interferon-induced protein 4;ADAR1 adenosine deaminase RNA -

(MDR/TAP), Member 1 ABL1 NM 00
Official Symbol Accession Official Full Name ABCB1 NM_000927.3 ATP-binding cassette, sub-family B (MDR/TAP), member 1 ABL1 NM_005157.3 c-abl oncogene 1, non-receptor tyrosine kinase ADA NM_000022.2 adenosine deaminase AHR NM_001621.3 aryl hydrocarbon receptor AICDA NM_020661.1 activation-induced cytidine deaminase AIRE NM_000383.2 autoimmune regulator APP NM_000484.3 amyloid beta (A4) precursor protein ARG1 NM_000045.2 arginase, liver ARG2 NM_001172.3 arginase, type II ARHGDIB NM_001175.4 Rho GDP dissociation inhibitor (GDI) beta ATG10 NM_001131028.1 ATG10 autophagy related 10 homolog (S. cerevisiae) ATG12 NM_004707.2 ATG12 autophagy related 12 homolog (S. cerevisiae) ATG16L1 NM_198890.2 ATG16 autophagy related 16-like 1 (S. cerevisiae) ATG5 NM_004849.2 ATG5 autophagy related 5 homolog (S. cerevisiae) ATG7 NM_001136031.2 ATG7 autophagy related 7 homolog (S. cerevisiae) ATM NM_000051.3 ataxia telangiectasia mutated B2M NM_004048.2 beta-2-microglobulin B3GAT1 NM_018644.3 beta-1,3-glucuronyltransferase 1 (glucuronosyltransferase P) BATF NM_006399.3 basic leucine zipper transcription factor, ATF-like BATF3 NM_018664.2 basic leucine zipper transcription factor, ATF-like 3 BAX NM_138761.3 BCL2-associated X protein BCAP31 NM_005745.7 B-cell receptor-associated protein 31 BCL10 NM_003921.2 B-cell CLL/lymphoma 10 BCL2 NM_000657.2 B-cell CLL/lymphoma 2 BCL2L11 NM_138621.4 BCL2-like 11 (apoptosis facilitator) BCL3 NM_005178.2 B-cell CLL/lymphoma 3 BCL6 NM_001706.2 B-cell CLL/lymphoma 6 BID NM_001196.2 BH3 interacting domain death agonist BLNK NM_013314.2 -

Transcriptomic Analysis Reveals Priming of the Host Antiviral
International Journal of Molecular Sciences Article Transcriptomic Analysis Reveals Priming of The Host Antiviral Interferon Signaling Pathway by Bronchobini® Resulting in Balanced Immune Response to Rhinovirus Infection in Mouse Lung Tissue Slices Stella Marie Reamon-Buettner 1 , Monika Niehof 1 , Natalie Hirth 2, Olga Danov 1 , Helena Obernolte 1, Armin Braun 1, Jürgen Warnecke 2, Katherina Sewald 1 and Sabine Wronski 1,* 1 Department of Preclinical Pharmacology and In Vitro Toxicology, Fraunhofer Institute for Toxicology and Experimental Medicine, Biomedical Research in Endstage and Obstructive Lung Disease Hannover (BREATH), Member of the German Center for Lung Research (DZL), Member of Fraunhofer international Consortium for Anti-Infective Research (iCAIR), 30625 Hannover, Germany; [email protected] (S.M.R.-B.); [email protected] (M.N.); [email protected] (O.D.); [email protected] (H.O.); [email protected] (A.B.); [email protected] (K.S.) 2 Heel GmbH, 76532 Baden-Baden, Germany; [email protected] (N.H.); [email protected] (J.W.) * Correspondence: [email protected]; Tel.: +49-511-5350-444 Received: 26 March 2019; Accepted: 30 April 2019; Published: 7 May 2019 Abstract: Rhinovirus (RV) is the predominant virus causing respiratory tract infections. Bronchobini® is a low dose multi component, multi target preparation used to treat inflammatory respiratory diseases such as the common cold, described to ease severity of symptoms such as cough and viscous mucus production. The aim of the study was to assess the efficacy of Bronchobini® in RV infection and to elucidate its mode of action.