Review of Lambda Interferons in Hepatitis B Virus Infection: Outcomes and Therapeutic Strategies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interleukin 28 Is a Potential Therapeutic Target for Sepsis
Clinical Immunology 205 (2019) 29–34 Contents lists available at ScienceDirect Clinical Immunology journal homepage: www.elsevier.com/locate/yclim Interleukin 28 is a potential therapeutic target for sepsis T ⁎ Qin Luoa,b, Yi Liuc, Shuang Liub, Yibing Yinb, Banglao Xud, Ju Caoa, a Department of Laboratory Medicine, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China b Key Laboratory of Diagnostic Medicine designated by the Ministry of Education, Chongqing Medical University, Chongqing, China c Department of Intensive Care Unit, The Second Affiliated Hospital of Chongqing Medical University, Chongqing, China d Department of Laboratory Medicine, Guangzhou First People's Hospital, School of Medicine, South China University of Technology, Guangzhou, Guangdong, China ARTICLE INFO ABSTRACT Keywords: Identification of new therapeutic targets for the treatment of sepsis is imperative. We report here that cytokine Interleukin-28 IL-28 (IFN-λ) levels were elevated in clinical and experimental sepsis. Neutralization of IL-28 protected mice Sepsis from lethal sepsis induced by cecal ligation and puncture (CLP), which was associated with improved bacterial Infection clearance and enhanced neutrophil infiltration. Conversely, administration of recombinant IL-28 aggravated Immunity mortality, facilitated bacterial dissimilation and limited neutrophil recruitment, in the model of sepsis induced Neutrophil by CLP. This study defines IL-28 as a detrimental mediator during sepsis and identifies a potential therapeutic target for the immune therapy in sepsis. 1. Introduction immunopathology of sepsis is still poorly understood. To address this issue, we examined the potential role of IL-28 in the Each year, about 31.5 million individuals develop sepsis, and up to progression of sepsis. -

Mechanism of Action Through an IFN Type I-Independent Responses To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology , 12 of which you can access for free at: 2012; 188:3088-3098; Prepublished online 20 from submission to initial decision 4 weeks from acceptance to publication February 2012; doi: 10.4049/jimmunol.1101764 http://www.jimmunol.org/content/188/7/3088 MF59 and Pam3CSK4 Boost Adaptive Responses to Influenza Subunit Vaccine through an IFN Type I-Independent Mechanism of Action Elena Caproni, Elaine Tritto, Mario Cortese, Alessandro Muzzi, Flaviana Mosca, Elisabetta Monaci, Barbara Baudner, Anja Seubert and Ennio De Gregorio J Immunol cites 33 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/02/21/jimmunol.110176 4.DC1 This article http://www.jimmunol.org/content/188/7/3088.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 -

MOUSE INTERLEUKIN-28B/INTERFERON-LAMBDA 3, CARRIER FREE Product Number: 12821-1 Lot Number: 5559 Size: 25 Μg
MOUSE INTERLEUKIN-28B/INTERFERON-LAMBDA 3, CARRIER FREE Product Number: 12821-1 Lot Number: 5559 Size: 25 µg Description: Mouse Interleukin-28B/Interferon-lambda 3, Carrier Free Source: A DNA sequence encoding the mature mouse IL-28B/IFN-λ3 (Asp 20 - Val 193) (Kotenko, S.V. et al., 2003, Nat. Immunol. 4(1):69 - 77) was expressed in E. coli. Form: Lyophilized Buffer: Phosphate-buffered saline (PBS) Reconstitution: It is recommended that sterile PBS be added to the vial to prepare a stock solution of no less than 100 μg/mL. Endotoxin: < 1 EU/µg Molecular Weight: The 175 amino acid residue methionyl form of recombinant mouse IL-28B has a predicted molecular mass of approximately 19.7 kDa. Purity: > 95% Synonyms: Mu IL-28B; Mu IFN-λ3 Accession #: NP_796370 Assays Used to Measure Bioactivity: Human HepG2 cells infected with encephalomyocarditis virus (Sheppard, P. et al., 2003, Nature Immunol. 4:63). The ED50 for this effect is typically 7.5 - 37.5 ng/mL. Shipping Conditions: Wet Ice Physical State of Product During Shipping: Lyophilized Special Conditions/Comments: After receipt, this product should be kept at -70˚C or below for retention of full activity. Upon reconstitution, this cytokine can be stored under sterile conditions at 2˚C to 8˚C for one month or at -20˚C to -70˚C in a manual defrost freezer for three months without detection loss of activity. Avoid repeated freeze-thaw cycles. For more information on protein handling, visit the PBL website at www.interferonsource.com . Product Information: Human IL-28A, IL-28B, and IL-29, also named interferon-λ2 (IFN-λ2), IFN-λ3, and IFN-λ1, respectively, are newly identified class II cytokine receptor ligands that are distantly related to members of the IL-10 family (11- 13% aa sequence identity) and type I IFN family (15 - 19% aa sequence identity).1 – 3 The genes encoding these three cytokines are localized to chromosome 19 and each is composed of multiple exons. -

Interplay Between Hepatitis D Virus and the Interferon Response
viruses Review Interplay between Hepatitis D Virus and the Interferon Response Zhenfeng Zhang 1 and Stephan Urban 1,2,* 1 Department of Infectious Diseases, Molecular Virology, University Hospital Heidelberg, 69120 Heidelberg, Germany; [email protected] 2 German Centre for Infection Research (DZIF), Partner Site Heidelberg, 69120 Heidelberg, Germany * Correspondence: [email protected]; Tel.: +49-6221-564-902 Received: 27 October 2020; Accepted: 18 November 2020; Published: 20 November 2020 Abstract: Chronic hepatitis D (CHD) is the most severe form of viral hepatitis, with rapid progression of liver-related diseases and high rates of development of hepatocellular carcinoma. The causative agent, hepatitis D virus (HDV), contains a small (approximately 1.7 kb) highly self-pairing single-strand circular RNA genome that assembles with the HDV antigen to form a ribonucleoprotein (RNP) complex. HDV depends on hepatitis B virus (HBV) envelope proteins for envelopment and de novo hepatocyte entry; however, its intracellular RNA replication is autonomous. In addition, HDV can amplify HBV independently through cell division. Cellular innate immune responses, mainly interferon (IFN) response, are crucial for controlling invading viruses, while viruses counteract these responses to favor their propagation. In contrast to HBV, HDV activates profound IFN response through the melanoma differentiation antigen 5 (MDA5) pathway. This cellular response efficiently suppresses cell-division-mediated HDV spread and, to some extent, early stages of HDV de novo infection, but only marginally impairs RNA replication in resting hepatocytes. In this review, we summarize the current knowledge on HDV structure, replication, and persistence and subsequently focus on the interplay between HDV and IFN response, including IFN activation, sensing, antiviral effects, and viral countermeasures. -

The Role of Il-29 and Il-28B in the Innate Immune Response
THE ROLE OF IL-29 AND IL-28B IN THE INNATE IMMUNE RESPONSE Megumi A. Williamson A thesis submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Master of Science in the Department of Periodontology, School of Dentistry Chapel Hill 2018 Approved by: Thiago Morelli Thiago Morelli Julie Marchesan Steven Offenbacher Antonio Amelio ©2018 Megumi A. Williamson ALL RIGHTS RESERVED ii ABSTRACT Megumi A. Williamson: The Role of IL-29 and IL-28B in the Innate Immune Response (Under the direction of Thiago Morelli, Julie Marchesan, Steven Offenbacher, and Antonio Amelio) Aims: Chronic periodontitis (CP) is an inflammatory disease induced by dysbiotic biofilm in a susceptible host, resulting in progressive attachment loss, and subsequent alveolar bone loss. Recent genome-wide association studies (GWAS) and genome-wide gene centric analysis on periodontal complex traits (PCTs) identified possible associations of IL-29 and IL- 28B with periodontal diseases. However, the underlying mechanisms for how these genes contribute to the pathogenesis of periodontitis are largely unknown. The aims of the present study were to explore the role of IL-29 and IL-28B and their gene polymorphisms in the innate immune response by dendritic cells. Materials and methods: To explore the effect of IL-29 on the cytokine production in response to TLR4 stimulation, the IL-29 gene was knocked-down in THP-1 cells using IL-29 shRNA lentiviral particles. Pro- and anti-inflammatory cytokine levels were measured with Luminex® multiplex assay. To assess the effect of genetic variations in IL- 29 and IL-28B, whole blood samples from fifteen subjects (6 subjects with major allele for both IL-29 and IL-28B, 5 subjects for major allele in IL-29 and minor allele in IL-28B, and 4 subjects with minor alleles for both genes) were collected and CD14+/CD16lo PBMCs were isolated to generate DCs. -

Estimating the Long-Term Clinical and Economic Outcomes of Daclatasvir
VALUE IN HEALTH REGIONAL ISSUES ] ( ]]]]) ]]]– ]]] Available online at www.sciencedirect.com journal homepage: www.elsevier.com/locate/vhri Estimating the Long-Term Clinical and Economic Outcomes of Daclatasvir Plus Asunaprevir in Difficult-to-Treat Japanese Patients Chronically Infected with Hepatitis C Genotype 1b Phil McEwan, PhD1,2, Thomas Ward, MSc1,*, Samantha Webster, BSc1, Yong Yuan, PhD3, Anupama Kalsekar, MS3, Kristine Broglio, MS4, Isao Kamae, PhD5, Melanie Quintana, PhD4, Scott M. Berry, PhD4, Masahiro Kobayashi, MD6, Sachie Inoue, PhD7, Ann Tang, PhD8, Hiromitsu Kumada, MD6 1Health Economics and Outcomes Research Ltd., Monmouth, UK; 2Centre for Health Economics, Swansea University, Swansea, UK; 3Global Health Economics and Outcomes Research, Princeton, NJ, USA; 4Berry Consultants, LLC, Austin, TX, USA; 5Graduate School of Public Policy, University of Tokyo, Tokyo, Japan; 6Department of Hepatology, Toranomon Hospital, Tokyo, Japan; 7CRECON Research and Consulting, Inc., Tokyo, Japan; 8Health Economics and Outcomes Research, Bristol-Myers K.K., Tokyo, Japan ABSTRACT Objectives: Japan has one of the highest endemic rates of hepatitis C treated with TVR þ pegIFN-α/RBV. Results: Initiating DCV þ ASV virus (HCV) infection. Treatments in Japan are currently limited to treatment in patients in the chronic hepatitis C disease stage resulted interferon-alfa–based regimens, which are associated with tolerability in quality-adjusted life-year gains of 0.96 and 0.77 over TVR þ pegIFN- and efficacy issues. A novel regimen combining two oral HCV α/RBV for NRs and PRs, respectively, and a gain of 2.61 in interferon- therapies, daclatasvir and asunaprevir (DCV þ ASV), has shown alfa–ineligible/intolerant patients over no treatment. -

Cytokines: from Basic Mechanisms of Control to New Therapeutics
This is a free sample of content from Cytokines: From Basic Mechanisms of Control to New Therapeutics. Click here for more information on how to buy the book. Index A Cryopyrin-associated periodic syndromes (CAPS), AKT 262–263 g chain family cytokine signaling, 9 CTCL. See Cutaneous T-cell lymphoma T-cell development role, 279 Cutaneous T-cell lymphoma (CTCL), interleukin-15 role, AMPs. See Antimicrobial peptides 448 Antimicrobial peptides (AMPs), 456 Cytokine release syndrome (CRS), interleukin-6 therapeutic Arid5a, 94 targeting, 93, 94 Atopic dermatitis Cytokine studies interleukin-17 studies, 130 agonist development, 458–459 interleukin-22 therapeutic targeting, 169 alarmins, 456–457 antagonist development, 459–460 assay development, 457 B definition, 455–456 B cell historical perspective, 456 g chain family cytokine function, 16–18 mimics, 457 interferon-g in activation, 226–227 mutation studies, 458 tumor necrosis factor in development and function, 105 signaling studies, 457–458 BCL11B, 280–281 b Common family cytokines. See also specific cytokines cancer studies, 46 D functions, 43–44 Daclizumab, 21 nervous system function, 46–47 DC. See Dendritic cell overview, 40–43 Dendritic cell (DC) receptors classical dendritic cells activation, 48–49, 51 cDC1 development and function, 397–398 assembly, 48 cDC2 development and function, 398–399 assembly, 48–51 GM-DCs, 403–404 interactions with other receptors, 47–48 human cell features, 401–402 proximal activation of signaling, 52–53 identification sepsis and inflammation studies, 44–45 mouse bone marrow, 396–397 therapy, 41–42, 53–56 surface markers for in vivo identification, 403 BLIMP1, 15 maturation, 404–406 Briakinumab, 151 Mo-DCs, 403 g chain family cytokine function, 19 C interferon-g in activation, 226 Candida albicans, interleukin-17 studies, 129 long noncoding RNA regulation of differentiation, CAPS. -

Molecular Characterization of Tea Catechin Treated Human Prostate Cancer Cell Lines Yewseok Suh
Florida State University Libraries Electronic Theses, Treatises and Dissertations The Graduate School 2006 Molecular Characterization of Tea Catechin Treated Human Prostate Cancer Cell Lines Yewseok Suh Follow this and additional works at the FSU Digital Library. For more information, please contact [email protected] THE FLORIDA STATE UNIVERSITY COLLEGE OF ARTS AND SCIENCES MOLECULAR CHARACTERIZATION OF TEA CATECHIN TREATED HUMAN PROSTATE CANCER CELL LINES By YEWSEOK SUH A Dissertation submitted to the Department of Chemistry and Biochemistry in partial fulfillment of the requirements for the degree of Doctor of Philosophy Degree Awarded: Summer Semester, 2006 The members of the Committee approve the dissertation of Yewseok Suh defended on May 12, 2006. Qing-Xiang Amy Sang Professor Directing Dissertation Thomas C.S. Keller III Outside Committee Member Joseph B. Schlenoff Committee Member Hong Li Committee Member Approved: Naresh Dalal, Chair, Department of Chemistry and Biochemistry Joseph Travis, Dean, College of Arts and Sciences The Office of Graduate Studies has verified and approved the above named committee members. ii This dissertation is dedicated to my parents for their endless love and encouragement, to my lovely wife Inok Park for her support, and to our charming daughter, Tae-won. iii ACKNOWLEDGEMENTS I would like to express my gratitude to my major professor for her support throughout the research. Also thanks to all my committee members, Dr. Thomas C. S. Keller III, Dr. Hong Li, and Dr. Joseph B. Schlenoff, for their support, advice, and guidance. Special thanks to our lab members especially, Ziad Sahab and Robert G. Newcomer and former lab member Douglas R. -

2018 Chen Lingyan 1448129
This electronic thesis or dissertation has been downloaded from the King’s Research Portal at https://kclpure.kcl.ac.uk/portal/ Genetics and Epigenetics in Systemic Lupus Erythematosus Chen, Lingyan Awarding institution: King's College London The copyright of this thesis rests with the author and no quotation from it or information derived from it may be published without proper acknowledgement. END USER LICENCE AGREEMENT Unless another licence is stated on the immediately following page this work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International licence. https://creativecommons.org/licenses/by-nc-nd/4.0/ You are free to copy, distribute and transmit the work Under the following conditions: Attribution: You must attribute the work in the manner specified by the author (but not in any way that suggests that they endorse you or your use of the work). Non Commercial: You may not use this work for commercial purposes. No Derivative Works - You may not alter, transform, or build upon this work. Any of these conditions can be waived if you receive permission from the author. Your fair dealings and other rights are in no way affected by the above. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 03. Oct. 2021 GENETICS AND EPIGENETICS IN SYSTEMIC LUPUS ERYTHEMATOSUS Lingyan CHEN Department of Medical & Molecular Genetics Faculty of Life Sciences & Medicine King’s College London This thesis is submitted for the degree of DoCtor of Philosophy (PhD) 1 To my parents 2 DeClaration The work described in this thesis was carried out within the Vyse Immunogenetics Group in the Department of Medical and Molecular Genetics at King’s College London under the supervision of Professor Timothy James Vyse and Dr David Lester Morris and between the years of October 2014 and March 2018. -
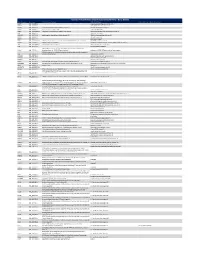
Ncounter® Autoimmune Discovery Consortium Panel
nCounter® Autoimmune Discovery Consortium Panel - Gene Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information AAMP NM_001087.3 angio associated migratory cell protein ABHD6 NM_020676.5 abhydrolase domain containing 6 ACKR2 NM_001296.3 CMKBR9,CCBP2;chemokine binding protein 2 atypical chemokine receptor 2 ACOXL NM_018308.1 acyl-Coenzyme A oxidase-like acyl-CoA oxidase like ACSL6 NM_001009185.1 FACL6;fatty-acid-Coenzyme A ligase, long-chain 6 acyl-CoA synthetase long chain family member 6 ADA NM_000022.2 adenosine deaminase ADAM30 NM_021794.2 a disintegrin and metalloproteinase domain 30 ADAM metallopeptidase domain 30 ADCY3 NM_004036.3 adenylate cyclase 3 ADCY7 NM_001114.4 adenylate cyclase 7 AFF3 NM_001025108.1 LAF4;lymphoid nuclear protein related to AF4,AF4/FMR2 family, member 3 AF4/FMR2 family member 3 AGAP2 NM_014770.3 CENTG1;centaurin, gamma 1 ArfGAP with GTPase domain, ankyrin repeat and PH domain 2 AHI1 NM_001134830.1 Abelson helper integration site Abelson helper integration site 1 AHR NM_001621.3 aryl hydrocarbon receptor AHSA2;AHA1, activator of heat shock 90kDa protein ATPase homolog 2 AHSA2 NM_152392.1 (yeast),activator of HSP90 ATPase homolog 2 activator of HSP90 ATPase homolog 2, pseudogene APECED;autoimmune regulator (autoimmune polyendocrinopathy candidiasis AIRE NM_000383.2 ectodermal dystrophy) autoimmune regulator AMIGO3 NM_198722.2 adhesion molecule with Ig like domain 3 ANKRD55 NM_024669.2 ankyrin repeat domain 55 ANTXR2 NM_058172.5 anthrax toxin receptor -

S Sofosbuvir (Sovaldi )
Sofosbuvir (Sovaldi ™) UTILIZATION MANAGEMENT CRITERIA DRUG CLASS: Nucleotide Analog NS5B Polymerase Inhibitor BRAND (generic) NAME: Sovaldi (sofosbuvir ) 400 mg strength tablet FDA -APPROVED INDICATIONS Sovaldi is a hepatitis C virus (HCV) nucleotide analog NS5B polymerase inhibitor indicated for the treatment of chronic hepatitis C (CHC) infection as a component of a combination antiviral treatment regimen. • Sovaldi efficacy has been established in subjects with HCV genotype 1, 2, 3 or 4 infection, including those with hepatocellular carcinoma meeting Milan criteria (awaiting liver transplantation) and those with HCV/HIV -1 co-infection. COVERAGE AUTHORIZATION CRITE RIA Sofosbuvir (Sovaldi) is cover ed for the following condition and situations: • Diagnosis of chronic hepatitis C (CHC) infection with c onfirmed genotype 1, 2, 3, or 4, OR • CHC infection with hepatocellular carcinoma awaiting liver transplant, AND • Sofosbuvir is prescribed in combination with ribavirin and pegylated interferon alfa for genotypes 1 and 4, OR • Sofosbuvir is prescribed in combination with ribavirin for p atients with genotype 1 who are peginterferon-ineligible* (medical record documentation of ineligibility for peginterferon therapy must be submitted for review), OR • Sofosbuvir is prescribed in combination with simeprevir, with or without ribavirin, for 12 weeks duration of therapy, for patients with genotype 1 that have advanced fi brosis (METAVIR score 2, 3, 4, OR cirrhosis secondary to Hepatitis C ), and are peginterferon - ineligible* (medical record -

Pegasys®) Peginterferon Alfa-2B (Peg-Intron®
Peginterferon alfa-2a (Pegasys®) Peginterferon alfa-2b (Peg-Intron®) UTILIZATION MANAGEMENT CRITERIA DRUG CLASS: Pegylated Interferons BRAND (generic) NAME: Pegasys (peginterferon alfa-2a) Single-use vial 180 mcg/1.0 mL; Prefilled syringe 180 mcg/0.5 mL Peg-Intron (peginterferon alfa-2b) Single-use vial (with 1.25 mL diluent) and REDIPEN®: 50 mcg, 80 mcg, 120 mcg, 150 mcg per 0.5 mL. FDA-APPROVED INDICATIONS PEG-Intron (peginterferon alfa-2b): Combination therapy with ribavirin: • Chronic Hepatitis C (CHC) in patients 3 years of age and older with compensated liver disease. • Patients with the following characteristics are less likely to benefit from re-treatment after failing a course of therapy: previous nonresponse, previous pegylated interferon treatment, significant bridging fibrosis or cirrhosis, and genotype 1 infection. Monotherapy: CHC in patients (18 years of age and older) with compensated liver disease previously untreated with interferon alpha. Pegasys (Peginterferon alfa-2a): • Treatment of Chronic Hepatitis C (CHC) in adults with compensated liver disease not previously treated with interferon alpha, in patients with histological evidence of cirrhosis and compensated liver disease, and in adults with CHC/HIV coinfection and CD4 count > 100 cells/mm3. o Combination therapy with ribavirin is recommended unless patient has contraindication to or significant intolerance to ribavirin. Pegasys monotherapy is indicated for: • Treatment of adult patients with HBeAg positive and HBeAg negative chronic hepatitis B who have compensated