PQS Ab Initio Program Package Version 3.3
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Semiempirical Implementations of Molecular
Semiempirical Implementations of Molecular Orbital Theory How one can make Hartree-Fock theory less computationally intensive without much sacrificing its accuracy? The most demanding step – calculation of two-electron (four-index) integrals (J and K integrals) appearing in the Fock matrix elements (N4 where N is the number of basis functions). One way to save time – to estimate their value accurately in an a priori fashion and thus to avoid numerical integration. Coulomb integrals measure the repulsion between electrons in regions of space defined by the basis functions. When the basis functions in the integral for one electron are very far from the basis functions for the other, the value of that integral will approach zero. In a large molecule, one might be able to avoid the calculation of a very large number of integrals simply by assuming them to be zero. HF theory is intrinsically inaccurate as it does not include correlation energy. Therefore, modifications of the theory introduced in order to simplify its formalism may actually improve it, provided the new approximations somehow introduce an accounting for correlation energy. Most typically, such approximations involve the adoption of a parametric form for some aspect of the calculation where the parameters involved are chosen so as best reproduce experimental data – ‘semiempirical’. Another motivation for introducing semiempirical approximation into HF theory was to facilitate the computation of derivatives (gradients, Hessians) so that geometries could be more efficiently optimized. Extended Hückel Theory Before considering semiempirical methods we revisit Hückel theory: H11 − ES11 H12 − ES12 L H1N − ES1N H21 − ES21 H22 − ES22 L H2N − ES2N = 0 M M O M H − ES H − ES H − ES N1 N1 N2 N 2 L NN NN The dimension of the secular determinant depends on the choice of the basis set. -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -
![Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021](https://docslib.b-cdn.net/cover/7378/automated-construction-of-quantum-classical-hybrid-models-arxiv-2102-09355v1-physics-chem-ph-18-feb-2021-177378.webp)
Automated Construction of Quantum–Classical Hybrid Models Arxiv:2102.09355V1 [Physics.Chem-Ph] 18 Feb 2021
Automated construction of quantum{classical hybrid models Christoph Brunken and Markus Reiher∗ ETH Z¨urich, Laboratorium f¨urPhysikalische Chemie, Vladimir-Prelog-Weg 2, 8093 Z¨urich, Switzerland February 18, 2021 Abstract We present a protocol for the fully automated construction of quantum mechanical-(QM){ classical hybrid models by extending our previously reported approach on self-parametri- zing system-focused atomistic models (SFAM) [J. Chem. Theory Comput. 2020, 16 (3), 1646{1665]. In this QM/SFAM approach, the size and composition of the QM region is evaluated in an automated manner based on first principles so that the hybrid model describes the atomic forces in the center of the QM region accurately. This entails the au- tomated construction and evaluation of differently sized QM regions with a bearable com- putational overhead that needs to be paid for automated validation procedures. Applying SFAM for the classical part of the model eliminates any dependence on pre-existing pa- rameters due to its system-focused quantum mechanically derived parametrization. Hence, QM/SFAM is capable of delivering a high fidelity and complete automation. Furthermore, since SFAM parameters are generated for the whole system, our ansatz allows for a con- venient re-definition of the QM region during a molecular exploration. For this purpose, a local re-parametrization scheme is introduced, which efficiently generates additional clas- sical parameters on the fly when new covalent bonds are formed (or broken) and moved to the classical region. arXiv:2102.09355v1 [physics.chem-ph] 18 Feb 2021 ∗Corresponding author; e-mail: [email protected] 1 1 Introduction In contrast to most protocols of computational quantum chemistry that consider isolated molecules, chemical processes can take place in a vast variety of complex environments. -

Semiempirical Quantum-Chemical Methods Max-Planck-Institut Für
Max-Planck-Institut für Kohlenforschung This is the peer reviewed version of the following article: WIREs Comput. Mol. Sci. 4, 145-157 (2014), which has been published in final form at https://doi.org/10.1002/wcms.1161. This article may be used for non-commercial purposes in accordance with Wiley Terms and Conditions for Use of Self- Archived Versions. Semiempirical quantum-chemical methods Walter Thiel Max-Planck-Institut für Kohlenforschung Kaiser-Wilhelm-Platz 1, 45470 Mülheim, Germany [email protected] Abstract The semiempirical methods of quantum chemistry are reviewed, with emphasis on established NDDO-based methods (MNDO, AM1, PM3) and on the more recent orthogonalization-corrected methods (OM1, OM2, OM3). After a brief historical overview, the methodology is presented in non- technical terms, covering the underlying concepts, parameterization strategies, and computational aspects, as well as linear scaling and hybrid approaches. The application section addresses selected recent benchmarks and surveys ground-state and excited-state studies, including recent OM2- based excited-state dynamics investigations. Introduction Quantum mechanics provides the conceptual framework for understanding chemistry and the theoretical foundation for computational methods that model the electronic structure of chemical compounds. There are three types of such approaches: Quantum-chemical ab initio methods provide a convergent path to the exact solution of the Schrödinger equation and can therefore give “the right answer for the right reason”, but they are costly and thus restricted to relatively small molecules (at least in the case of the highly accurate correlated approaches). Density functional theory (DFT) has become the workhorse of computational chemistry because of its favourable price/performance ratio, allowing for fairly accurate calculations on medium-size molecules, but there is no systematic path of improvement in spite of the first-principles character of DFT. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
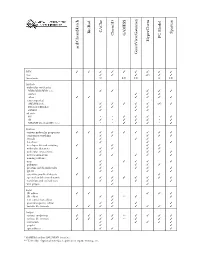
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Dmol Guide to Select a Dmol3 Task 1
DMOL3 GUIDE MATERIALS STUDIO 8.0 Copyright Notice ©2014 Dassault Systèmes. All rights reserved. 3DEXPERIENCE, the Compass icon and the 3DS logo, CATIA, SOLIDWORKS, ENOVIA, DELMIA, SIMULIA, GEOVIA, EXALEAD, 3D VIA, BIOVIA and NETVIBES are commercial trademarks or registered trademarks of Dassault Systèmes or its subsidiaries in the U.S. and/or other countries. All other trademarks are owned by their respective owners. Use of any Dassault Systèmes or its subsidiaries trademarks is subject to their express written approval. Acknowledgments and References To print photographs or files of computational results (figures and/or data) obtained using BIOVIA software, acknowledge the source in an appropriate format. For example: "Computational results obtained using software programs from Dassault Systèmes Biovia Corp.. The ab initio calculations were performed with the DMol3 program, and graphical displays generated with Materials Studio." BIOVIA may grant permission to republish or reprint its copyrighted materials. Requests should be submitted to BIOVIA Support, either through electronic mail to [email protected], or in writing to: BIOVIA Support 5005 Wateridge Vista Drive, San Diego, CA 92121 USA Contents DMol3 1 Setting up a molecular dynamics calculation20 Introduction 1 Choosing an ensemble 21 Further Information 1 Defining the time step 21 Tasks in DMol3 2 Defining the thermostat control 21 Energy 3 Constraints during dynamics 21 Setting up the calculation 3 Setting up a transition state calculation 22 Dynamics 4 Which method to use? -
![Modern Quantum Chemistry with [Open]Molcas](https://docslib.b-cdn.net/cover/4742/modern-quantum-chemistry-with-open-molcas-744742.webp)
Modern Quantum Chemistry with [Open]Molcas
Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 Submitted: 17 February 2020 . Accepted: 11 May 2020 . Published Online: 05 June 2020 Francesco Aquilante , Jochen Autschbach , Alberto Baiardi , Stefano Battaglia , Veniamin A. Borin , Liviu F. Chibotaru , Irene Conti , Luca De Vico , Mickaël Delcey , Ignacio Fdez. Galván , Nicolas Ferré , Leon Freitag , Marco Garavelli , Xuejun Gong , Stefan Knecht , Ernst D. Larsson , Roland Lindh , Marcus Lundberg , Per Åke Malmqvist , Artur Nenov , Jesper Norell , Michael Odelius , Massimo Olivucci , Thomas B. Pedersen , Laura Pedraza-González , Quan M. Phung , Kristine Pierloot , Markus Reiher , Igor Schapiro , Javier Segarra-Martí , Francesco Segatta , Luis Seijo , Saumik Sen , Dumitru-Claudiu Sergentu , Christopher J. Stein , Liviu Ungur , Morgane Vacher , Alessio Valentini , and Valera Veryazov J. Chem. Phys. 152, 214117 (2020); https://doi.org/10.1063/5.0004835 152, 214117 © 2020 Author(s). The Journal ARTICLE of Chemical Physics scitation.org/journal/jcp Modern quantum chemistry with [Open]Molcas Cite as: J. Chem. Phys. 152, 214117 (2020); doi: 10.1063/5.0004835 Submitted: 17 February 2020 • Accepted: 11 May 2020 • Published Online: 5 June 2020 Francesco Aquilante,1,a) Jochen Autschbach,2,b) Alberto Baiardi,3,c) Stefano Battaglia,4,d) Veniamin A. Borin,5,e) Liviu F. Chibotaru,6,f) Irene Conti,7,g) Luca De Vico,8,h) Mickaël Delcey,9,i) Ignacio Fdez. Galván,4,j) Nicolas Ferré,10,k) Leon Freitag,3,l) Marco Garavelli,7,m) Xuejun Gong,11,n) Stefan Knecht,3,o) Ernst D. Larsson,12,p) Roland Lindh,4,q) Marcus Lundberg,9,r) Per Åke Malmqvist,12,s) Artur Nenov,7,t) Jesper Norell,13,u) Michael Odelius,13,v) Massimo Olivucci,8,14,w) Thomas B. -

Computational Studies on Carbohydrates: I. Density Functional Ab Initio Geometry Optimization on Maltose Conformations
Computational Studies on Carbohydrates: I. Density Functional Ab Initio Geometry Optimization on Maltose Conformations F. A. MOMANY, J. L. WILLETT Plant Polymer Research, National Center for Agricultural Utilization Research, USDA, Agricultural Research Service, 1815 N. University St., Peoria, Illinois 61604 Received 10 September 1999; accepted 10 May 2000 ABSTRACT: Ab initio geometry optimization was carried out on 10 selected conformations of maltose and two 2-methoxytetrahydropyran conformations using the density functional denoted B3LYP combined with two basis sets. The 6-31G∗ and 6-311CCG∗∗ basis sets make up the B3LYP/6-31G∗ and B3LYP/6-311CCG∗∗ procedures. Internal coordinates were fully relaxed, and structures were gradient optimized at both levels of theory. Ten conformations were studied at the B3LYP/6-31G∗ level, and five of these were continued with ∗∗ full gradient optimization at the B3LYP/6-311CCG level of theory. The details of the ab initio optimized geometries are presented here, with particular attention given to the positions of the atoms around the anomeric center and the effect of the particular anomer and hydrogen bonding pattern on the maltose ring structures and relative conformational energies. The size and complexity of the hydrogen-bonding network prevented a rigorous search of conformational space by ab initio calculations. However, using empirical force fields, low-energy conformers of maltose were found that were subsequently gradient optimized at the two ab initio levels of theory. Three classes of conformations were studied, as defined by the clockwise or counterclockwise direction of the hydroxyl groups, or a flipped conformer in which the -dihedral is rotated by ∼180◦.Different combinations of ! side-chain rotations gave energy differences of more than 6 kcal/mol above the lowest energy structure found. -

Quantum Mechanical Calculation of Molybdenum and Tungsten Influence on the Crm-Oxide Catalyst Acidity Oyegoke Toyese1 Fadimatu N
Hittite Journal of Science and Engineering, 2020, 7 (4) 297–311 ISSN NUMBER: 2148–4171 DOI: 10.17350/HJSE19030000199 Quantum Mechanical Calculation of Molybdenum and Tungsten Influence on the CrM-oxide Catalyst Acidity Oyegoke Toyese1 Fadimatu N. Dabai1 Adamu Uzairur2 and Baba El-Yakubu Jibril1 1Ahmadu Bello University, Department of Chemical Engineering, Zaria, Nigeria 2Ahmadu Bello University, Department of Chemistry, Zaria, Nigeria Article History: ABSTRACT Received: 2020/06/18 Accepted: 2020/11/02 Online: 2020/12/31 emi-empirical calculations were employed to understand the effects of introducing pro- Smoters such as molybdenum (Mo) and tungsten (W) on chromium (III) oxide catalyst Correspondence to: Oyegoke Toyese, for the dehydrogenation of propane into propylene. For this purpose, we investigated CrM- Ahmadu Bello University, Chemical Engineering, Zaria, NIGERIA oxide (M = Cr, Mo, and W) catalysts. In this study, the Lewis acidity of the catalyst was ex- E-Mail: [email protected] amined using Lewis acidity parameters (Ac), including ammonia and pyridine adsorption Phone: +234 703 047 91 06 energy. The results obtained from this study of overall acidity across all sites of the catalysts studied reveal Mo-modified catalyst as the one with the least acidity while the W-modified catalyst was found to have shown the highest acidity signifies that the introduction of Mo would reduce acidity while W accelerates it. The finding, therefore, confirms tungsten (W) to be more influential and would be more promising when compared to molybdenum (Mo) due to the better avenue that is offered by W for the promotion of electron exchange and its higher acidity(s).