Hypervalent Radioactive Astatine Or Iodine Compounds, and Preparation Methods Thereof
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

FRANCIUM Element Symbol: Fr Atomic Number: 87
FRANCIUM Element Symbol: Fr Atomic Number: 87 An initiative of IYC 2011 brought to you by the RACI KAYE GREEN www.raci.org.au FRANCIUM Element symbol: Fr Atomic number: 87 Francium (previously known as eka-cesium and actinium K) is a radioactive metal and the second rarest naturally occurring element after Astatine. It is the least stable of the first 103 elements. Very little is known of the physical and chemical properties of Francium compared to other elements. Francium was discovered by Marguerite Perey of the Curie Institute in Paris, France in 1939. However, the existence of an element of atomic number 87 was predicted in the 1870s by Dmitri Mendeleev, creator of the first version of the periodic table, who presumed it would have chemical and physical properties similar to Cesium. Several research teams attempted to isolate this missing element, and there were at least four false claims of discovery during which it was named Russium (after the home country of soviet chemist D. K. Dobroserdov), Alkalinium (by English chemists Gerald J. K. Druce and Frederick H. Loring as the heaviest alkali metal), Virginium (after Virginia, home state of chemist Fred Allison), and Moldavium (by Horia Hulubei and Yvette Cauchois after Moldavia, the Romanian province where they conducted their work). Perey finally discovered Francium after purifying radioactive Actinium-227 from Lanthanum, and detecting particles decaying at low energy levels not previously identified. The new product exhibited chemical properties of an alkali metal (such as co-precipitating with Cesium salts), which led Perey to believe that it was element 87, caused by the alpha radioactive decay of Actinium-227. -

Microdosimetry of Astatine-211 and Xa0102708 Comparison with That of Iodine-125
MICRODOSIMETRY OF ASTATINE-211 AND XA0102708 COMPARISON WITH THAT OF IODINE-125 T. UNAK Ege University, Bomova, Izmir, Turkey Abstract.' 21lAt is an alpha and Auger emitter radionuclide and has been frequently used for labeling of different kind of chemical agents. 125I is also known as an effective Auger emitter. The radionuclides which emit short range and high LET radiations such as alpha particles and Auger electrons have high radiotoxic effectiveness on the living systems. The microdosimetric data are suitable to clarify the real radiotoxic effectiveness and to get the detail of diagnostic and therapeutic application principles of these radionuclides. In this study, the energy and dose absorptions by cell nucleus from alpha particles and Auger electrons emitted by 211At have been calculated using a Monte Carlo calculation program (code: UNMOC). For these calculations two different model corresponding to the cell nucleus have been used and the data obtained were compared with the data earlier obtained for I25I. As a result, the radiotoxicity of 21lAt is in the competition with 125I. In the case of a specific agent labelled with 2I1At or 125I is incorporated into the cell or cell nucleus, but non-bound to DNA or not found very close to it, 2llAt should considerably be much more radiotoxic than i25I, but in the case of the labelled agent is bound to DNA or take a place very close to it, the radiotoxicity of i25I should considerably be higher than 21'At. 1. INTRODUCTION 21'At as an alpha and Auger emitter radionuclide has an extremely high potential application in cancer therapy; particularly, in molecular radiotherapy. -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

Enigmatic Astatine D
in your element Enigmatic astatine D. Scott Wilbur points out the difficulty in studying the transient element astatine, and the need to understand its basic chemical nature to help in the development of targeted radiotherapy agents. ince the discovery of astatine The other ‘long’-lived isotope, 210At, is not of labelling reagents containing more over 70 years ago1, many of its suitable because it decays into polonium-210 stable aromatic astatine–boron bonds Scharacteristics have remained elusive. — the notorious radiation poison used to has improved that situation, and studies Unlike the other halogens, abundant and kill the Russian Federal Security Service evaluating bonding with other elements ubiquitous in nature, astatine is one of officer Alexander Litvinenko in 2006, may further advance it. the rarest of all elements. This arises from after he took political asylum in the To determine in vivo stability, the the fact that it has no stable isotopes; the United Kingdom. same cancer-targeting molecule can longest lived of its 32 known radioisotopes, Although some chemical data has be labelled with 211At and (stably) with 210At, has a half-life of only 8.1 hours. been compiled for astatine isotopes, radioiodine (125I, 123I or 131I), and the two The rarity and radioactive nature of many physical properties have only been co-injected. The concentrations of 211At element 85 lends to its mystery, as it cannot extrapolated. Similar to other halogens, in various tissues (higher lung, spleen, be observed or weighed in a conventional astatine undergoes nucleophilic and stomach and thyroid) indicate whether it is sense. Even its colour is unknown; based electrophilic reactions. -
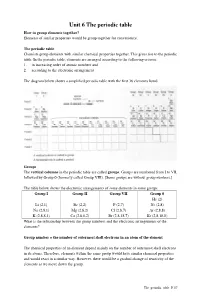
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -

ASTATINE Element Symbol: at Atomic Number: 85
ASTATINE Element Symbol: At Atomic Number: 85 An initiative of IYC 2011 brought to you by the RACI LINDA ABBLITT www.raci.org.au ASTATINE Element symbol: At Atomic number: 85 Astatine is a highly radioactive chemical element. It is chemically similar to the other halogens above it in Group 17 of the periodic table. It is the heaviest known halogen. As chemists would expect, Astatine acts more like a metal than iodine, the element just above it in the table. Astatine is produced by radioactive decay in nature, but due to its short half-life it is found only in minute amounts. It is currently the rarest naturally occurring element, with less than 30 grams estimated to be contained in the entire Earth’s crust. This amounts to less than one teaspoon of the element. Isaac Asimov in a 1957 essay on large numbers, scientific notation and the size of the atom, wrote that in “all of North and South America to a depth of ten miles”, the number of astatine-215 atoms at any time is “only a trillion”. Guinness Book of Records lists it as the rarest element. It is found near thorium and uranium in the Earth’s crust. Astatine would be expected to be a nearly black solid, which, when heated, sublimes into a dark, purplish vapor (darker than iodine) Astatine (after Greek astatos meaning unstable) was first synthesized in 1940 by Dale R Corson, Kenneth Ross MacKenzie an Emilio Segrè at the University of California, Berkeley by bombarding bismuth with alpha particles in a cyclotron. -

Exploration of Astatine Chemistry in Solution : Focus on the Pourbaix Diagram in Noncomplexing Medium and Characterization of Astatine-Mediated Halogen Bonds Lu Liu
Exploration of astatine chemistry in solution : focus on the Pourbaix diagram in noncomplexing medium and characterization of astatine-mediated halogen bonds Lu Liu To cite this version: Lu Liu. Exploration of astatine chemistry in solution : focus on the Pourbaix diagram in noncom- plexing medium and characterization of astatine-mediated halogen bonds. Radiochemistry. Ecole na- tionale supérieure Mines-Télécom Atlantique, 2020. English. NNT : 2020IMTA0205. tel-03123005 HAL Id: tel-03123005 https://tel.archives-ouvertes.fr/tel-03123005 Submitted on 27 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THESE DE DOCTORAT DE L’ÉCOLE NATIONALE SUPERIEURE MINES-TELECOM ATLANTIQUE BRETAGNE PAYS DE LA LOIRE - IMT ATLANTIQUE ECOLE DOCTORALE N° 596 Matière, Molécules, Matériaux Spécialité : Chimie Analytique et Radiochimie Par Lu LIU Exploration de la chimie de l'astate en solution : Focalisation sur le diagramme de Pourbaix en milieu non complexant et caractérisation de liaisons halogènes induites par l'astate Thèse présentée et soutenue à Nantes, -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

Condensed Astatine: Monatomic and Metallic
week ending PRL 111, 116404 (2013) PHYSICAL REVIEW LETTERS 13 SEPTEMBER 2013 Condensed Astatine: Monatomic and Metallic Andreas Hermann School of Physics and Astronomy and Centre for Science at Extreme Conditions, University of Edinburgh, Edinburgh, EH9 3JZ, United Kingdom Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA Roald Hoffmann Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA N. W. Ashcroft Laboratory of Atomic and Solid State Physics, Cornell University, Ithaca, New York 14853, USA (Received 10 May 2013; published 12 September 2013) The condensed matter properties of the nominal terminating element of the halogen group with atomic number 85, astatine, are as yet unknown. In the intervening more than 70 years since its discovery significant advances have been made in substrate cooling and the other techniques necessary for the production of the element to the point where we might now enquire about the key properties astatine might have if it attained a condensed phase. This subject is addressed here using density functional theory and structural selection methods, with an accounting for relativistic physics that is essential. Condensed astatine is predicted to be quite different in fascinating ways from iodine, being already at 1 atm a metal, and monatomic at that, and possibly a superconductor (as is dense iodine). DOI: 10.1103/PhysRevLett.111.116404 PACS numbers: 71.20. b, 71.15.Mb, 71.15.Rf À When examining Mendeleev’s enduring periodic array to prevent deterioration of a macroscopic sample of with respect to the physical attributes of the condensed astatine. -

Food Periodic Table
Food Chemistry Periodic Table Celebrating the International Year of the Periodic Table 2019 Created by Jane K Parker Acknowledgements and thanks to the RSC Food Group Committee: Robert Cordina, Bryan Hanley, Taichi Inuit, John Points, Kathy Ridgway, Martin Rose, Wendy Russell, Mike Saltmarsh, Maud Silvent, Clive Thomson, Kath Whittaker, Pete Wilde, and to Martin Chadwick, Cian Moloney and Ese Omoaruhke, for contributions to the elements, to Flaticons for use of their free icons, and to Alinea and TDMA for photographs of He and Ti. In slideshow mode, click on an element in periodic table to find out more, return via the RSC Food Group Logo. Contact: [email protected] Food Chemistry Periodic Table H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Cs Ba La Hf Ta W Re Os Ir Pt Au Hg Ti Pb Bi Po At Rn Fr Ra Ac Rf Db Sg Bh Hs Mt Ds Rg Cn Nh Fl Mc Lv Ts Og H Hydrogen 1 Occurrences in food Roles in food • Core element in organic compounds (fats, proteins, • H+ gives one of the five basic tastes – sour. carbohydrates, vitamins). • the higher the concentration of H+, the lower the pH • OH pH 2 Lemon juice (very sour) • H2 H2 H2 H2 H2 pH 3 Apple (sour) C C C C C C • pH 5 Meat (not sour) H3C C C C C C O H H H H H 2 2 2 2 2 • pH 7 Tea or water (not sour) • Hydrogen-bonds, one of the strongest forms of bonding, are crucial for the 3D-structure of many • Key element in water, H2O, which is 70% of the human body and 70% of many foods.