Exploration of Astatine Chemistry in Solution : Focus on the Pourbaix Diagram in Noncomplexing Medium and Characterization of Astatine-Mediated Halogen Bonds Lu Liu
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

FRANCIUM Element Symbol: Fr Atomic Number: 87
FRANCIUM Element Symbol: Fr Atomic Number: 87 An initiative of IYC 2011 brought to you by the RACI KAYE GREEN www.raci.org.au FRANCIUM Element symbol: Fr Atomic number: 87 Francium (previously known as eka-cesium and actinium K) is a radioactive metal and the second rarest naturally occurring element after Astatine. It is the least stable of the first 103 elements. Very little is known of the physical and chemical properties of Francium compared to other elements. Francium was discovered by Marguerite Perey of the Curie Institute in Paris, France in 1939. However, the existence of an element of atomic number 87 was predicted in the 1870s by Dmitri Mendeleev, creator of the first version of the periodic table, who presumed it would have chemical and physical properties similar to Cesium. Several research teams attempted to isolate this missing element, and there were at least four false claims of discovery during which it was named Russium (after the home country of soviet chemist D. K. Dobroserdov), Alkalinium (by English chemists Gerald J. K. Druce and Frederick H. Loring as the heaviest alkali metal), Virginium (after Virginia, home state of chemist Fred Allison), and Moldavium (by Horia Hulubei and Yvette Cauchois after Moldavia, the Romanian province where they conducted their work). Perey finally discovered Francium after purifying radioactive Actinium-227 from Lanthanum, and detecting particles decaying at low energy levels not previously identified. The new product exhibited chemical properties of an alkali metal (such as co-precipitating with Cesium salts), which led Perey to believe that it was element 87, caused by the alpha radioactive decay of Actinium-227. -

Microdosimetry of Astatine-211 and Xa0102708 Comparison with That of Iodine-125
MICRODOSIMETRY OF ASTATINE-211 AND XA0102708 COMPARISON WITH THAT OF IODINE-125 T. UNAK Ege University, Bomova, Izmir, Turkey Abstract.' 21lAt is an alpha and Auger emitter radionuclide and has been frequently used for labeling of different kind of chemical agents. 125I is also known as an effective Auger emitter. The radionuclides which emit short range and high LET radiations such as alpha particles and Auger electrons have high radiotoxic effectiveness on the living systems. The microdosimetric data are suitable to clarify the real radiotoxic effectiveness and to get the detail of diagnostic and therapeutic application principles of these radionuclides. In this study, the energy and dose absorptions by cell nucleus from alpha particles and Auger electrons emitted by 211At have been calculated using a Monte Carlo calculation program (code: UNMOC). For these calculations two different model corresponding to the cell nucleus have been used and the data obtained were compared with the data earlier obtained for I25I. As a result, the radiotoxicity of 21lAt is in the competition with 125I. In the case of a specific agent labelled with 2I1At or 125I is incorporated into the cell or cell nucleus, but non-bound to DNA or not found very close to it, 2llAt should considerably be much more radiotoxic than i25I, but in the case of the labelled agent is bound to DNA or take a place very close to it, the radiotoxicity of i25I should considerably be higher than 21'At. 1. INTRODUCTION 21'At as an alpha and Auger emitter radionuclide has an extremely high potential application in cancer therapy; particularly, in molecular radiotherapy. -

© Copyright 2016 Devon R. Mortensen
© Copyright 2016 Devon R. Mortensen Understanding near Fermi-level electronic structure through x-ray emission spectroscopy Devon R. Mortensen A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2016 Reading Committee: Gerald T. Seidler, Chair Marjorie Olmstead Xiaodong Xu Program Authorized to Offer Degree: Physics University of Washington Abstract Understanding near Fermi-level electronic structure through x-ray emission spectroscopy Devon R. Mortensen Chair of the Supervisory Committee: Professor Gerald T. Seidler Physics Atomic and molecular chemical properties are largely determined by the electronic structure of near Fermi-level states. Determining this structure is therefore one of the central tasks in materials characterization and development. In the work of this dissertation I explore the capabilities and limitations of non-resonant x-ray emission spectroscopy (NXES) as an analytical technique aimed at addressing these issues. To this end, I report the development of novel laboratory- and synchrotron-based instrumentation for the study of transition metal and lanthanide compounds. One of the primary results of this research thrust is increased accessibility and throughput, making NXES measurements a more viable option in routine and research-grade materials study. Using experimental data obtained from these spectrometers, I evaluate current state-of-the-art theory in terms of modeling valence structure in ambient transition-metal complexes. Additionally, I use NXES to elucidate the evolving 4f-electronic structure in the early light lanthanides under pressure. In particular these results show a persistent 4f-moment across certain volume collapse transitions in Cerium and Praseodymium, thus helping settle a long-standing debate about the nature of volume collapse. -

New Technologies for 211At Targeted Α-Therapy Research Using 211Rn and 209At
New technologies for 211At targeted α-therapy research using 211Rn and 209At Jason Raymond Crawford BSc, University of British Columbia, 2007 MSc, University of Victoria, 2010 A dissertation submitted in partial fulfilment of the requirements for the degree of Doctorate of Philosophy in the Department of Physics and Astronomy c Jason Raymond Crawford, 2016 University of Victoria All rights reserved. This dissertation may not be reproduced in whole or in part by photocopy or other means, without the permission of the author. ii New technologies for 211At targeted α-therapy research using 211Rn and 209At by Jason Raymond Crawford BSc, University of British Columbia, 2007 MSc, University of Victoria, 2010 Supervisory Committee Dr. Thomas J Ruth, Co-Supervisor Department of Physics and Astronomy Dr. Andrew Jirasek, Co-Supervisor Department of Physics and Astronomy Dr. Wayne Beckham, Committee Member Department of Physics and Astronomy Dr. Dean Karlen, Committee Member Department of Physics and Astronomy Dr. Julian Lum, Outside Member Department of Biochemistry & Microbiology iii Abstract The most promising applications for targeted α-therapy with astatine-211 (211At) in- clude treatments of disseminated microscopic disease, the major medical problem for cancer treatment. The primary advantages of targeted α-therapy with 211At are that the α-particle radiation is densely ionizing, translating to high relative biological effectiveness (RBE), and short-range, minimizing damage to surrounding healthy tissues. In addition, theranostic imaging with 123I surrogates has shown promise for developing new therapies with 211At and translating them to the clinic. Currently, Canada does not have a way of producing 211At by conventional methods because it lacks α-particle accelerators with necessary beam energy and intensity. -

Of the Periodic Table
of the Periodic Table teacher notes Give your students a visual introduction to the families of the periodic table! This product includes eight mini- posters, one for each of the element families on the main group of the periodic table: Alkali Metals, Alkaline Earth Metals, Boron/Aluminum Group (Icosagens), Carbon Group (Crystallogens), Nitrogen Group (Pnictogens), Oxygen Group (Chalcogens), Halogens, and Noble Gases. The mini-posters give overview information about the family as well as a visual of where on the periodic table the family is located and a diagram of an atom of that family highlighting the number of valence electrons. Also included is the student packet, which is broken into the eight families and asks for specific information that students will find on the mini-posters. The students are also directed to color each family with a specific color on the blank graphic organizer at the end of their packet and they go to the fantastic interactive table at www.periodictable.com to learn even more about the elements in each family. Furthermore, there is a section for students to conduct their own research on the element of hydrogen, which does not belong to a family. When I use this activity, I print two of each mini-poster in color (pages 8 through 15 of this file), laminate them, and lay them on a big table. I have students work in partners to read about each family, one at a time, and complete that section of the student packet (pages 16 through 21 of this file). When they finish, they bring the mini-poster back to the table for another group to use. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title PART A. AN X-RAY SPECTROMETER FOR USE IN RADIOACTIVITY MEASUREMENTS, THE L X- RAYS OF NEPTUNIUM AND PLUTONIUM. PART B. SOME LIGHTER ISOTOPES OF ASTATINE Permalink https://escholarship.org/uc/item/9ss952mr Author Barton, G.W. Publication Date 1950-05-02 eScholarship.org Powered by the California Digital Library University of California ·. l '; UCRL~67o · cy 2 UNIVERSITY OF CALIFORNIA TWO-WEEK LOAN COPY This is a Library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Division, Ext. 5545 BERKELEY, CALIFORNIA DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. • .\ UCRL-670 No Distribution II]ECLASSIF~ED UNIVERSITY OF CALIFORNIA Radiation Laboratory Contract No. -

Experimental Aspects of Geoneutrino Detection: Status and Perspectives
Experimental Aspects of Geoneutrino Detection: Status and Perspectives O. Smirnov,1 1JINR, Joint Institute for Nuclear Research, Dubna, Russian Federation October 22, 2019 Abstract Neutrino geophysics, the study of the Earth's interior by measuring the fluxes of geologically produced neutrino at its surface, is a new interdisciplinary field of science, rapidly developing as a synergy between geology, geophysics and particle physics. Geoneutrinos, antineutrinos from long- lived natural isotopes responsible for the radiogenic heat flux, provide valuable information for the chemical composition models of the Earth. The calculations of the expected geoneutrino signal are discussed, together with experimental aspects of geoneutrino detection, including the description of possible backgrounds and methods for their suppression. At present, only two detectors, Borexino and KamLAND, have reached sensitivity to the geoneutrino. The experiments accumulated a set of ∼190 geoneutrino events and continue the data acquisition. The detailed description of the experiments, their results on geoneutrino detection, and impact on geophysics are presented. The start of operation of other detectors sensitive to geoneutrinos is planned for the near future: the SNO+ detector is being filled with liquid scintillator, and the biggest ever 20 kt JUNO detector is under construction. A review of the physics potential of these experiments with respect to the geoneutrino studies, along with other proposals, is presented. New ideas and methods for geoneutrino detection are reviewed. Contents 1 Introduction 2 2 Geoneutrinos and the Earth's heat 4 2.1 Long-lived radiogenic elements . 4 2.2 Radiogenic heat and geoneutrino luminosity of the Earth . 9 arXiv:1910.09321v1 [physics.geo-ph] 21 Oct 2019 3 Geoneutrino flux calculation 11 3.1 Neutrino oscillations . -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title PRODUCTION AND DECAY PROPERTIES OF THORIUM ISOTOPES OF MASS 221-224 FORMED IN HEAVY ION REACTIONS Permalink https://escholarship.org/uc/item/9bp2f39g Authors Valli, Kalevi Hyde, Earl K. Borggreen, Jorn. Publication Date 1969-10-01 eScholarship.org Powered by the California Digital Library University of California Submitted to Physical Review UCRL-18992 Preprint ,. 2 "'--' Ht:::.cE.l'VED l'VJRENCE RAD\AliOI'l lABORAlOR'l tEt3 Z6 1970 ..,. , L\BRARY AND N ""NTS sE.CTlO .1 0 ocut'J\r;;.. PRODUCTION AND DECAY PROPERTIES OF THORIUM ISOTOPES p• OF MASS 221-224 FORMED IN HEAVY ION REACTIONS Kalevi Valli, Earl K. Hyde, and Jprn Borggreen October 1969 AEC Contract No. W -7405-eng-48 TWO-WEEK LOAN COPY This is a library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call ··~~. Tech. Info. Division, Ext. 5545 ,.... -< () LAWRENCE RADIATION LABORATOR~ Z ~0~ UNI,1ERSI'"fY of C:AJ_.JIF'()RNIA BEllKELEry rt ·oce ' ' DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain coiTect information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any waiTanty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. -

Enigmatic Astatine D
in your element Enigmatic astatine D. Scott Wilbur points out the difficulty in studying the transient element astatine, and the need to understand its basic chemical nature to help in the development of targeted radiotherapy agents. ince the discovery of astatine The other ‘long’-lived isotope, 210At, is not of labelling reagents containing more over 70 years ago1, many of its suitable because it decays into polonium-210 stable aromatic astatine–boron bonds Scharacteristics have remained elusive. — the notorious radiation poison used to has improved that situation, and studies Unlike the other halogens, abundant and kill the Russian Federal Security Service evaluating bonding with other elements ubiquitous in nature, astatine is one of officer Alexander Litvinenko in 2006, may further advance it. the rarest of all elements. This arises from after he took political asylum in the To determine in vivo stability, the the fact that it has no stable isotopes; the United Kingdom. same cancer-targeting molecule can longest lived of its 32 known radioisotopes, Although some chemical data has be labelled with 211At and (stably) with 210At, has a half-life of only 8.1 hours. been compiled for astatine isotopes, radioiodine (125I, 123I or 131I), and the two The rarity and radioactive nature of many physical properties have only been co-injected. The concentrations of 211At element 85 lends to its mystery, as it cannot extrapolated. Similar to other halogens, in various tissues (higher lung, spleen, be observed or weighed in a conventional astatine undergoes nucleophilic and stomach and thyroid) indicate whether it is sense. Even its colour is unknown; based electrophilic reactions. -
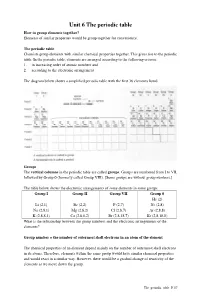
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

Annotated Bibliography: Women in Physics, Astronomy, and Related Disciplines
Annotated Bibliography: Women in Physics, Astronomy, and Related Disciplines Abir Am, Pnina and Dorinda Outram, eds. Uneasy Careers and Intimate Lives: Women in Science, 1787-1979. New Brunswick, NJ: Rutgers University Press, 1987. Abir Am and Outram’s volume includes a collection of essays about women in science that highlight the intersection of personal and professional spheres. All of the articles argue that the careers of women scientists are influenced by their family lives and that their family lives are impacted because of their scientific careers. This text is significant in two ways: first, it is one of the earliest examples of scholarship that moves beyond the recovering women in science, but placing them in the context of their home and work environments. Second, it suggests that historians of science can no longer ignore the private lives of their historical subjects. This volume contains four articles relating to women in physics and astronomy: Marilyn Bailey Ogilvie’s “Marital Collaboration: An Approach to Science” (pages 104-125), Sally Gregory Kohlstedt’s “Maria Mitchell and the Advancement of Women in Science” (pages 129-146), Helena M. Pycior’s “Marie Curie’s ‘Anti-Natural Path’: Time Only for Science and Family” (pages 191-215), and Peggy Kidwell’s “Cecelia Payne-Gaposchkin: Astronomy in the Family” (pages 216-238). As a unit, the articles would constitute and interesting lesson on personal and professional influences. Individually, the articles could be incorporated into lessons on a single scientist, offering a new perspective on their activities at work and at home. It complements Pycior, Slack, and Abir Am’s Creative Couples in the Sciences and Lykknes, Opitz, and Van Tiggelen’s For Better of For Worse: Collaborative Couples in the Sciences, which also look at the intersection of the personal and professional.