The Structure of Oxalate Decarboxylase at Its Active Ph
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Proteomic Analysis of Mycelial Proteins from Magnaporthe Oryzae Under Nitrogen Starvation
Proteomic analysis of mycelial proteins from Magnaporthe oryzae under nitrogen starvation X.-G. Zhou1,2, P. Yu 2, C. Dong2, C.-X. Yao2, Y.-M. Ding2, N. Tao2 and Z.-W. Zhao1 1State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan, Yunnan University, Kunming, China 2Key Laboratory of Southwestern Crop Gene Resources and Germplasm Innovation, Ministry of Agriculture and Yunnan Provincial Key Laboratory of Agricultural Biotechnology, Biotechnology and Germplasm Resources Institute, Yunnan Academy of Agricultural Sciences, Kunming, China Corresponding author: Z.W. Zhao E-mail: [email protected] Genet. Mol. Res. 15 (2): gmr.15028637 Received March 23, 2016 Accepted April 11, 2016 Published May 13, 2016 DOI http://dx.doi.org/10.4238/gmr.15028637 ABSTRACT. Magnaporthe oryzae is an important model system in studies of plant pathogenic fungi, and nitrogen is a key nutrient source affecting microbial growth and development. In order to understand how nitrogen stress causes changes in mycelial proteins, we analyzed differentially expressed mycelial proteins from the M. oryzae virulent strain CH-63 using two-dimensional electrophoresis and mass spectrometry in complete medium or under nitrogen starvation conditions. A total of 975 ± 70 and 1169 ± 90 protein spots were detected in complete medium and under nitrogen starvation conditions, respectively. Forty-nine protein spots exhibited at least 2-fold up- regulation or down-regulation at the protein level according to PDQuest7.4. Moreover, 43 protein spots were successfully identified by matrix-assisted laser desorption/ionization-time-of-flight/time-of-flight mass spectrometry. Among these spots, 6 proteins were functionally unknown and 37 proteins were categorized into 5 groups according to Genetics and Molecular Research 15 (2): gmr.15028637 ©FUNPEC-RP www.funpecrp.com.br X.-G. -

Oxdc Antibody Rabbit Polyclonal Antibody Catalog # ABV11223
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 OxdC Antibody Rabbit Polyclonal Antibody Catalog # ABV11223 Specification OxdC Antibody - Product Information Application WB Primary Accession O34714 Reactivity Human Host Rabbit Clonality Polyclonal Isotype Rabbit IgG Calculated MW 43566 OxdC Antibody - Additional Information Gene ID 938620 Positive Control Western Blot: Recombinant protein Application & Usage Western blot: 1-4 Western blot of Oxalate decarboxylase µg/ml. antibody. Lane 1: rb- Oxalate decarboxylase - Other Names 10 ng. Lane 2: rb- Oxalate decarboxylase - 50 YvrK ng Target/Specificity OxdC OxdC Antibody - Background Antibody Form Oxalate decarboxylase (OxdC, EC4.1.1.2) is a Liquid manganese-containing enzyme, which decomposes oxalic acid and oxalate. With Appearance OxdC catalysis, oxalate is split into formate Colorless liquid and CO2. This enzyme belongs to the family of lyases, specifically the carboxy-lyases, which Formulation 100 µg (0.5 mg/ml) of antibody in PBS pH cleave carbon-carbon bonds. The systematic 7.2 containing 0.01 % BSA, 0.01 % name of this enzyme class is oxalate thimerosal, and 50 % glycerol. carboxy-lyase (formate-forming). This enzyme is also called oxalate carboxy-lyase. The Handling enzyme is composed of two cupin domains, The antibody solution should be gently each of which contains a Mn (II) ion. This mixed before use. enzyme participates in glyoxylate and dicarboxylate metabolism. This enzyme has Reconstitution & Storage been recognized for diagnostics in diverse -20 °C biotechnological applications such as the clinical assay of oxalate in blood and urine, Background Descriptions therapeutics, process industry, and agriculture to lower oxalate levels in foods and the environment. -

Proteo-Metabolomic Investigation of Transgenic Rice Unravels Metabolic
www.nature.com/scientificreports OPEN Proteo-metabolomic investigation of transgenic rice unravels metabolic alterations and Received: 27 November 2018 Accepted: 24 June 2019 accumulation of novel proteins Published: xx xx xxxx potentially involved in defence against Rhizoctonia solani Subhasis Karmakar1, Karabi Datta1, Kutubuddin Ali Molla2,3, Dipak Gayen4, Kaushik Das1, Sailendra Nath Sarkar1 & Swapan K. Datta1 The generation of sheath blight (ShB)-resistant transgenic rice plants through the expression of Arabidopsis NPR1 gene is a signifcant development for research in the feld of biotic stress. However, to our knowledge, regulation of the proteomic and metabolic networks in the ShB-resistant transgenic rice plants has not been studied. In the present investigation, the relative proteome and metabolome profles of the non–transformed wild-type and the AtNPR1-transgenic rice lines prior to and subsequent to the R. solani infection were investigated. Total proteins from wild type and transgenic plants were investigated using two-dimensional gel electrophoresis (2-DE) followed by mass spectrometry (MS). The metabolomics study indicated an increased abundance of various metabolites, which draws parallels with the proteomic analysis. Furthermore, the proteome data was cross-examined using network analysis which identifed modules that were rich in known as well as novel immunity-related prognostic proteins, particularly the mitogen-activated protein kinase 6, probable protein phosphatase 2C1, probable trehalose-phosphate phosphatase 2 and heat shock protein. A novel protein, 14–3– 3GF14f was observed to be upregulated in the leaves of the transgenic rice plants after ShB infection, and the possible mechanistic role of this protein in ShB resistance may be investigated further. -

Brno University of Technology Vysoké Učení Technické V Brně
BRNO UNIVERSITY OF TECHNOLOGY VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ FACULTY OF CHEMISTRY FAKULTA CHEMICKÁ INSTITUTE OF CHEMISTRY AND TECHNOLOGY OF ENVIRONMENTAL PROTECTION ÚSTAV CHEMIE A TECHNOLOGIE OCHRANY ŽIVOTNÍHO PROSTŘEDÍ DEGRADATION OF HEAT TRANSFER FLUIDS IN THERMAL SOLAR SYSTEMS AND PROPANE-1,3-DIOL AS A NEW OPTION STÁRNUTÍ TEPLONOSNÝCH KAPALIN V TERMICKÝCH SOLÁRNÍCH SYSTÉMECH A PROPAN-1,3-DIOL JAKO NOVÁ MOŽNOST DOCTORAL THESIS DIZERTAČNÍ PRÁCE AUTHOR Ing. František Mikšík AUTOR PRÁCE SUPERVISOR prof. Ing. Josef Čáslavský, CSc. ŠKOLITEL BRNO 2018 ABSTRAKT Stárnutí teplonosných kapalin na organické bázi je dlouhodobým problémem, který je znám od počátku jejich používání. První část této disertační práce je tak věnována případové studii funkčního experimentálního systému, který byl jako nový naplněn teplonosnou kapalinou na bázi propan-1,2-diol a pozorován po období 7 let. Pro analýzu stárnutí kapaliny v tomto systému byly sledovány základní provozní vlastnosti kapaliny jako jsou hustota, viskozita, teplota tuhnutí, pH a obsah kovů. Skrze tyto vlastnosti tak bylo sledováno stárnutí kapaliny nepřímo. Přímé sledování stárnutí bylo posléze provedeno analýzou degradačních produktů, jako jsou organické kyseliny a změny ve složení směsi pomocí izotachoforézy a hmotnostní spektrometrie. Pro srovnání byly taktéž analyzovány vybrané vzorky z několik dalších systémů plněných identickou kapalinou s prokazatelně pokročilou formou degradace. V druhé části práce jsou představeny základní fyzikálně-chemické vlastnosti směsí propan-1,3-diolu s vodou a jejich analytické hodnocení a matematické modelování pro universální použití jakožto nového základu pro nemrznoucí teplonosné kapaliny. Na základě dostupných informací je pak hodnocena použitelnost této směsi. Výhoda propan-1,3-diolu je spatřována především ve výrobě z obnovitelných zdrojů a v některých fyzikálních a chemických vlastnostech, které dle dosavadních poznatků předčívají doposud používané glykolové směsi. -

Oxalate Decarboxylase from Bacillus Subtilis, Recombinant Cat
Oxalate Decarboxylase from Bacillus subtilis, recombinant Cat. No. NATE-1688 Lot. No. (See product label) Introduction Description Oxalate decarboxylase (OxdC, EC4.1.1.2) is a manganese-containing enzyme, which decomposes oxalic acid and oxalate. With OxdC catalysis, oxalate is split into formate and CO2. This enzyme belongs to the family of lyases, specifically the carboxy-lyases, which cleave carbon-carbon bonds. The systematic name of this enzyme class is oxalate carboxy-lyase (formate-forming). This enzyme is also called oxalate carboxy-lyase. The enzyme is composed of two cupin domains, each of which contains a Mn (II) ion. This enzyme participates in glyoxylate and dicarboxylate metabolism. This enzyme has been recognized for diagnostics in diverse biotechnological applications such as the clinical assay of oxalate in blood and urine, therapeutics, process industry, and agriculture to lower oxalate levels in foods and the environment. The recombinant protein made from the Bacillus Subtilis sequence includes OxdC with N-terminal His-tag. Synonyms Oxalate carboxy-lyase; EC 4.1.1.2; Oxalate decarboxylase; OxdC Product Information Species Bacillus subtilis Source E. coli Form Liquid Formulation In 50 mM NaOAC, pH 5.5. The activation was stopped by addition of 10 mM MMTS, which can be removed under reducing conditions. EC Number EC 4.1.1.2 CAS No. 9024-97-9 Molecular Weight 45.9 kDa Purity > 98% by SDS-PAGE Activity 150U/mg Creative Enzymes. All rights reserved. 45-1 Ramsey Road, Shirley, NY 11967, USA Tel:1-631-562-8517 1-516-512-3133 Fax:1-631-938-8127 E-mail: [email protected] http://www.creative-enzymes.com Concentration 2 mg/mL Unit Definition One unit is the amount of enzyme that generates 1.0 µmole of NADH at 37°C. -

MECHANISM of the REACTION CATALYZED by the OXALATE DECARBOXYLASE from Bacillus Subtilis
MECHANISM OF THE REACTION CATALYZED BY THE OXALATE DECARBOXYLASE FROM Bacillus subtilis By DRAŽENKA SVEDRUŽIĆ A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 2005 Copyright 2005 by Draženka Svedružić This thesis is dedicated to my brother Željko, my parents and Chris. Ova je teza posvećena mome bratu Željku, mojim roditeljima i Krisu. ACKNOWLEDGMENTS This study was supported by grants from the National Institutes of Health (DK61666 and DK53556) and by the University of Florida Chemistry Department. Partial funding was also received from Dr. Ammon B. Peck. Thanks go to my doctoral dissertation committee: Dr. Steven A. Benner, Dr. Ammon B. Peck, Dr. Michael J. Scott, Dr. Jon D. Stewart, and especially my advisor, Dr. Nigel G. J. Richards, for making this research project a reality, but also for his constant support and guidance. Dr. Laurie A. Renhardt, Yang Liu and Dr. Wallace W. Cleland I thank for fruitful collaboration on heavy atom isotope effects research. Also, I thank Dr. Wallace W. Cleland for hospitality during my stay in Madison, Wisconsin. Thanks go to my EPR collaborators Dr. Lee Walker, Dr. Andrzej Ozarowski and Dr. Alexander Angerhofer. I am grateful to my coworkers and friends in the Richards research group, especially Dr. Christopher H. Chang for proofreading, countless discussions, valuable insights, guidance and support. Special thanks go to Stefan Jonsson for all his help, Sue Abbatiello for her mass spectrometry efforts and Lukas Koroniak for help with NMR experiments. Also thanks go to all members of Richards group, especially Mihai, Jemy and Cory, for providing a pleasant and supporting environment in and out of lab. -

(12) Patent Application Publication (10) Pub. No.: US 2009/0292100 A1 Fiene Et Al
US 20090292100A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0292100 A1 Fiene et al. (43) Pub. Date: Nov. 26, 2009 (54) PROCESS FOR PREPARING (86). PCT No.: PCT/EP07/57646 PENTAMETHYLENE 1.5-DIISOCYANATE S371 (c)(1), (75) Inventors: Martin Fiene, Niederkirchen (DE): (2), (4) Date: Jan. 9, 2009 (DE);Eckhard Wolfgang Stroefer, Siegel, Mannheim (30) Foreign ApplicationO O Priority Data Limburgerhof (DE); Stephan Aug. 1, 2006 (EP) .................................. O61182.56.4 Freyer, Neustadt (DE); Oskar Zelder, Speyer (DE); Gerhard Publication Classification Schulz, Bad Duerkheim (DE) (51) Int. Cl. Correspondence Address: CSG 18/00 (2006.01) OBLON, SPIVAK, MCCLELLAND MAIER & CD7C 263/2 (2006.01) NEUSTADT, L.L.P. CI2P I3/00 (2006.01) 194O DUKE STREET CD7C 263/10 (2006.01) ALEXANDRIA, VA 22314 (US) (52) U.S. Cl. ........... 528/85; 560/348; 435/128; 560/347; 560/355 (73) Assignee: BASFSE, LUDWIGSHAFEN (DE) (57) ABSTRACT (21) Appl. No.: 12/373,088 The present invention relates to a process for preparing pen tamethylene 1,5-diisocyanate, to pentamethylene 1,5-diiso (22) PCT Filed: Jul. 25, 2007 cyanate prepared in this way and to the use thereof. US 2009/0292100 A1 Nov. 26, 2009 PROCESS FOR PREPARING ene diisocyanates, especially pentamethylene 1,4-diisocyan PENTAMETHYLENE 1.5-DIISOCYANATE ate. Depending on its preparation, this proportion may be up to several % by weight. 0014. The pentamethylene 1,5-diisocyanate prepared in 0001. The present invention relates to a process for pre accordance with the invention has, in contrast, a proportion of paring pentamethylene 1,5-diisocyanate, to pentamethylene the branched pentamethylene diisocyanate isomers of in each 1.5-diisocyanate prepared in this way and to the use thereof. -

Comparative Studies of Oxalyl-Coa Decarboxylase Produced by Soil
3t' (O' COMPARATIVE STUDIES OF OXALYL.COA DECARBOXYLASE PRODUCED BY SOIL AND RUMINAL BACTERIA Thesis Submitted for the degree of Master of Agricultural Science in The University of Adelaide Faculty of Agricultural and Natural Resource Sciences by STEPHEN BOTTRILL November 1999 I I Table of Contents List of Figures VI List of Tables VM Abstract IX Acknowledgements XII Ståtement XIII List of Abbreviations XTV Chapær 1. Liærature Review 1 1.1 Introduction. 1 1.2 Exogenous Sources of Oxalates. 1 1.3 Endogenous Sources of Oxalate. 5 1.4 Poisoning. 9 1.4.1 Acute Poisoning" 10 1.4.2 Subacute Poisoning. 11 1.4.3 Chronic Poisoning. t2 1.4.4 SymPtoms in Humans. 14 1.4.5 Treatment of Poisoning. I4 1.4.6 Management to Prevent Poisoning. 15 1.5 Oxalate-Degrading Microorganisms. 18 1.6 Bacterial Classification 22 1.7 Pathways of Oxalate Degradation. 24 1.8 Formate in the Rumen. 27 1.9 Aims and Objectives. 29 Chapær 2. Materials and Methods 31 2.1 Materials 31 2.1.1 Chemicals 3r 2.1.2 EquiPment 31 2.I.3 Bacterial Strains and Plasmids 32 TI 2.1.4 Composition of Media 34 2.1.4.I Oxalate-Containing Media 34 2.1.4.I.1Liquid 34 2.1.4.1.2 Solid 34 2.1.4.2 O mlob act er formi g enes Media 35 2.I.4.2.I Trace Metals Solution 35 2.I.4.2.2 Medium A 35 2.1.4.2.3 Medium B 36 2.I.4.3 Luria-Bertani (LB) Broth 36 2.I.4.4 SOC Medium 37 2.2 Methods 37 2.2 -I Growth conditions 37 2.2.2 Isolation of oxal ate- de gradin g s oil bacteria 37 2.2.3 Characterisation of soil isolaæs 38 2.2.3.1 MicroscoPY 38 2.2.3.2 Gram stain 38 2.2.3.3 Carbon source utilisation 40 2.2.4.4 Volatile -

Structural and Mechanistic Studies on Α-Amino Β-Carboxymuconate Ε
Georgia State University ScholarWorks @ Georgia State University Chemistry Dissertations Department of Chemistry Summer 8-12-2014 Structural and Mechanistic Studies on α-Amino β- Carboxymuconate ε-Semialdehyde Decarboxylase and α- Aminomuconate ε-Semialdehyde Dehydrogenase Lu Huo Georgia State University Follow this and additional works at: https://scholarworks.gsu.edu/chemistry_diss Recommended Citation Huo, Lu, "Structural and Mechanistic Studies on α-Amino β-Carboxymuconate ε-Semialdehyde Decarboxylase and α-Aminomuconate ε-Semialdehyde Dehydrogenase." Dissertation, Georgia State University, 2014. https://scholarworks.gsu.edu/chemistry_diss/100 This Dissertation is brought to you for free and open access by the Department of Chemistry at ScholarWorks @ Georgia State University. It has been accepted for inclusion in Chemistry Dissertations by an authorized administrator of ScholarWorks @ Georgia State University. For more information, please contact [email protected]. STRUCTURAL AND MECHANISTIC STUDIES ON α-AMINO β-CARBOXYMUCONATE ε-SEMIALDEHYDE DECARBOXYLASE AND α-AMINOMUCONATE ε-SEMIALDEHYDE DEHYDROGENASE by LU HUO Under the Direction of Dr. Aimin Liu ABSTRACT α-Amino-β-carboxymuconate-ε-semialdehyde decarboxylase (ACMSD) and α- aminomuconate-ε-semialdehyde dehydrogenase (AMSDH) are two neighboring enzymes in the L-tryptophan and 2-nitrobenzoic acid degradation pathways. The substrates of the two enzymes, α-amino-β-carboxymuconate-ε-semialdehyde (ACMS) and α-aminomuconate-ε-semialdehyde (2-AMS), are unstable and spontaneously decay to quinolinic acid and picolinic acid, respectively. ACMSD utilizes a divalent zinc metal as cofactor and is a member of the amidohydrolase superfamily. In this dissertation work, we have identified an important histidine residue in the active site that plays dual roles in tuning metal selectivity and activating a metal bound water ligand using mutagenesis, resonance Raman, EPR, crystallography, and ICP metal analysis techniques. -

Crystal Structure of Oxalate Decarboxylase from Photorhabdus Luminescens, a Symbiotic Bacterium Associated with Entomopathogenic Nematodes
RESEARCH ARTICLES Crystal structure of oxalate decarboxylase from Photorhabdus luminescens, a symbiotic bacterium associated with entomopathogenic nematodes Sreeja Chellappan1,2, S. Mathivanan1, R. Thippeswamy3, M. Nagesh3, H. S. Savithri4 and M. R. N. Murthy1,5,6,* 1Molecular Biophysics Unit and 4Department of Biochemistry, Indian Institute of Science, Bengaluru 560 012, India 2Department of Molecular Biology, Kannur University, Kannur 671 314, India 3Indian Council of Agricultural Research, Project Directorate of Biological Control, Bengaluru 560 024, India 5Institute of Bioinformatics and Applied Biotechnology, Bengaluru 560 100, India 6Indian Institute of Science Education and Research, Thiruvananthapuram 695 551, India promotes nematode development and multiplication. Photorhabdus luminescens is a Gram-negative, symbi- 3 otic bacterium associated with entomopathogenic Duchaud et al. have determined the complete sequence nematodes of the genus Heterorhabditis. Several genes of P. luminescens. Analysis of the genomic sequence from this organism related to insecticidal properties revealed that it possesses a number of genes encoding for are being examined for their potential in pest man- potential virulence factors. agement. Oxalate decarboxylase (OXDC), an enzyme Oxalate decarboxylase (OXDC), proposed to have secreted by bacteria and fungi and putatively asso- insecticidal effect and secreted by bacteria, catalyses the ciated with insecticidal pathways catalyses the man- manganese-dependent decarboxylation of oxalate to ganese dependent decarboxylation of oxalate to formate and CO2. OXDC was first reported from fungi formate and CO2. In this study, we report the X-ray Flammulina (Collybia) velutipes and Coriolus hersutus4. crystal structure of OXDC isolated and purified from Later it was found in several other organisms5–12. OXDC Photorhabdus luminescens (PlOXDC, MW 43 kDa) structures have been determined from Bacillus subtilis determined at 1.97 Å resolution. -

Photorespiration Pathways in a Chemolithoautotroph
bioRxiv preprint doi: https://doi.org/10.1101/2020.05.08.083683; this version posted May 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Photorespiration pathways in a chemolithoautotroph Nico J. Claassens*1, Giovanni Scarinci*1, Axel Fischer1, Avi I. Flamholz2, William Newell1, Stefan Frielingsdorf3, Oliver Lenz3, Arren Bar-Even†1 1Max Planck Institute of Molecular Plant Physiology, Am Mühlenberg 1, 14476 Potsdam-Golm, Germany 2Department of Molecular and Cell Biology, University of California, Berkeley, California 94720, United States. 3Institut für Chemie, Physikalische Chemie, Technische Universität Berlin, Strasse des 17. Juni 135, 10623 Berlin, Germany †corresponding author; phone: +49 331 567-8910; Email: [email protected] *contributed equally Key words: CO2 fixation; hydrogen-oxidizing bacteria; glyoxylate shunt; malate synthase; oxalate metabolism 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.05.08.083683; this version posted May 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Abstract Carbon fixation via the Calvin cycle is constrained by the side activity of Rubisco with dioxygen, generating 2-phosphoglycolate. The metabolic recycling of 2-phosphoglycolate, an essential process termed photorespiration, was extensively studied in photoautotrophic organisms, including plants, algae, and cyanobacteria, but remains uncharacterized in chemolithoautotrophic bacteria. -
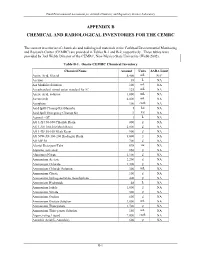
Appendix B Chemical and Radiological Inventories for the Cemrc
Final Environmental Assessment for Actinide Chemistry and Repository Science Laboratory APPENDIX B CHEMICAL AND RADIOLOGICAL INVENTORIES FOR THE CEMRC The current inventories of chemicals and radiological materials at the Carlsbad Environmental Monitoring and Research Center (CEMRC) are provided in Tables B-1 and B-2, respectively. These tables were provided by Joel Webb, Director of the CEMRC, New Mexico State University (Webb 2002). Table B-1. Onsite CEMRC Chemical Inventory Chemical Name Amount Units SARA Limit Acetic Acid, Glacial 5,400 mL NAa Acetone 38 L NA AA Modifier Solution 100 mL NA AccuStandard mixed anion standard for IC 125 mL NA Acetic Acid, solution 1,000 mL NA Acetonitrile 4,000 mL NA Acetylene 100 cu.ft. NA Acid Spill Cleanup Kit (Hazorb) 1 kit NA Acid Spill Emergency Cleanup Kit 3 kit NA Aerosol - OT 1 L NA AG 1-X4 50-100 Chloride Resin 800 g NA AG 1-X8 100-200 Mesh Resin 2,000 g NA AG 1-X8 50-100 Mesh Resin 900 g NA AG 50W-X8 100-200 Hydrogen Resin 3,000 g NA AG MP 50 700 g NA Alcojet Detergent/Tabs 698 oz NA Alumina, activated 850 g NA Aluminum Nitrate 2,100 g NA Ammonium Acetate 2,200 g NA Ammonium Chloride 1,300 g NA Ammonium Chloride Solution 300 mL NA Ammonium Citrate 100 g NA Ammonium hydrogenoxalate, hemihydrate 400 g NA Ammonium Hydroxide 48 L NA Ammonium Iodide 1,000 g NA Ammonium Nitrate 500 g NA Ammonium Oxalate 600 g NA Ammonium Oxalate Solution 2,000 mL NA Ammonium Thiocyanate 1,700 g NA Ammonium Thiocyanate Solution 150 mL NA Argon, refrig.