Characterization of Intermediate Oxidation States in CO2 Activation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brno University of Technology Vysoké Učení Technické V Brně
BRNO UNIVERSITY OF TECHNOLOGY VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ FACULTY OF CHEMISTRY FAKULTA CHEMICKÁ INSTITUTE OF CHEMISTRY AND TECHNOLOGY OF ENVIRONMENTAL PROTECTION ÚSTAV CHEMIE A TECHNOLOGIE OCHRANY ŽIVOTNÍHO PROSTŘEDÍ DEGRADATION OF HEAT TRANSFER FLUIDS IN THERMAL SOLAR SYSTEMS AND PROPANE-1,3-DIOL AS A NEW OPTION STÁRNUTÍ TEPLONOSNÝCH KAPALIN V TERMICKÝCH SOLÁRNÍCH SYSTÉMECH A PROPAN-1,3-DIOL JAKO NOVÁ MOŽNOST DOCTORAL THESIS DIZERTAČNÍ PRÁCE AUTHOR Ing. František Mikšík AUTOR PRÁCE SUPERVISOR prof. Ing. Josef Čáslavský, CSc. ŠKOLITEL BRNO 2018 ABSTRAKT Stárnutí teplonosných kapalin na organické bázi je dlouhodobým problémem, který je znám od počátku jejich používání. První část této disertační práce je tak věnována případové studii funkčního experimentálního systému, který byl jako nový naplněn teplonosnou kapalinou na bázi propan-1,2-diol a pozorován po období 7 let. Pro analýzu stárnutí kapaliny v tomto systému byly sledovány základní provozní vlastnosti kapaliny jako jsou hustota, viskozita, teplota tuhnutí, pH a obsah kovů. Skrze tyto vlastnosti tak bylo sledováno stárnutí kapaliny nepřímo. Přímé sledování stárnutí bylo posléze provedeno analýzou degradačních produktů, jako jsou organické kyseliny a změny ve složení směsi pomocí izotachoforézy a hmotnostní spektrometrie. Pro srovnání byly taktéž analyzovány vybrané vzorky z několik dalších systémů plněných identickou kapalinou s prokazatelně pokročilou formou degradace. V druhé části práce jsou představeny základní fyzikálně-chemické vlastnosti směsí propan-1,3-diolu s vodou a jejich analytické hodnocení a matematické modelování pro universální použití jakožto nového základu pro nemrznoucí teplonosné kapaliny. Na základě dostupných informací je pak hodnocena použitelnost této směsi. Výhoda propan-1,3-diolu je spatřována především ve výrobě z obnovitelných zdrojů a v některých fyzikálních a chemických vlastnostech, které dle dosavadních poznatků předčívají doposud používané glykolové směsi. -

The Structure of Oxalate Decarboxylase at Its Active Ph
bioRxivRunning preprint head: doi: https://doi.org/10.1101/426874 Low pH Structure, Oxalate; this version Decarboxylase posted September 25, 2018. The copyright holder for this preprint (which was1 not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The Structure of Oxalate Decarboxylase at its Active pH M. J. Burg, J. L. Goodsell, U. T. Twahir, S. D. Bruner, and A. Angerhofer, bioRxivLow preprintpH Structure, doi: https://doi.org/10.1101/426874 Oxalate Decarboxylase; this version posted September 25, 2018. The copyright holder for this preprint (which was2 not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Oxalate decarboxylase catalyzes the redox-neutral unimolecular disproportionation reaction of oxalic acid. The pH maximum for catalysis is ~4.0 and activity is negligible above pH7. Here we report on the first crystal structure of the enzyme in its active pH range at pH4.6, and at a resolution of 1.45 Å, the highest to date. The fundamental tertiary and quaternary structure of the enzyme does not change with pH. However, the low pH crystals are heterogeneous containing both a closed and open conformation of a flexible loop region which gates access to the N-terminal active site cavity. Residue E162 in the closed conformation points away from the active-site Mn ion owing to the coordination of a buffer molecule, acetate. Since the quaternary structure of the enzyme appears unaffected by pH many conclusions drawn from the structures taken at high pH remain valid. -
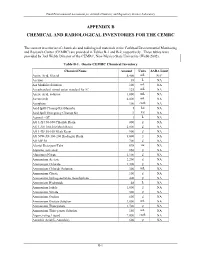
Appendix B Chemical and Radiological Inventories for the Cemrc
Final Environmental Assessment for Actinide Chemistry and Repository Science Laboratory APPENDIX B CHEMICAL AND RADIOLOGICAL INVENTORIES FOR THE CEMRC The current inventories of chemicals and radiological materials at the Carlsbad Environmental Monitoring and Research Center (CEMRC) are provided in Tables B-1 and B-2, respectively. These tables were provided by Joel Webb, Director of the CEMRC, New Mexico State University (Webb 2002). Table B-1. Onsite CEMRC Chemical Inventory Chemical Name Amount Units SARA Limit Acetic Acid, Glacial 5,400 mL NAa Acetone 38 L NA AA Modifier Solution 100 mL NA AccuStandard mixed anion standard for IC 125 mL NA Acetic Acid, solution 1,000 mL NA Acetonitrile 4,000 mL NA Acetylene 100 cu.ft. NA Acid Spill Cleanup Kit (Hazorb) 1 kit NA Acid Spill Emergency Cleanup Kit 3 kit NA Aerosol - OT 1 L NA AG 1-X4 50-100 Chloride Resin 800 g NA AG 1-X8 100-200 Mesh Resin 2,000 g NA AG 1-X8 50-100 Mesh Resin 900 g NA AG 50W-X8 100-200 Hydrogen Resin 3,000 g NA AG MP 50 700 g NA Alcojet Detergent/Tabs 698 oz NA Alumina, activated 850 g NA Aluminum Nitrate 2,100 g NA Ammonium Acetate 2,200 g NA Ammonium Chloride 1,300 g NA Ammonium Chloride Solution 300 mL NA Ammonium Citrate 100 g NA Ammonium hydrogenoxalate, hemihydrate 400 g NA Ammonium Hydroxide 48 L NA Ammonium Iodide 1,000 g NA Ammonium Nitrate 500 g NA Ammonium Oxalate 600 g NA Ammonium Oxalate Solution 2,000 mL NA Ammonium Thiocyanate 1,700 g NA Ammonium Thiocyanate Solution 150 mL NA Argon, refrig. -

Impurities Removal in Seawater to Optimize the Magnesium Extraction
IOP Conference Series: Materials Science and Engineering PAPER • OPEN ACCESS Related content - Fabrication and characterization of Impurities Removal in Seawater to Optimize the magnesium scaffold using different processing parameters Magnesium Extraction Saeid Toghyani and Mohammad Khodaei - Preparation of Superconducting Bi-Pb-Sr- Ca-Cu-O Compounds by Oxalate To cite this article: N C Natasha et al 2017 IOP Conf. Ser.: Mater. Sci. Eng. 176 012039 Coprecipitation Dian-Hau Chen, Cheng-Yie Shei, Shyang- Roeng Sheen et al. - Iron removal from a kaolinitic clay by leaching to obtain high whiteness index View the article online for updates and enhancements. R A Hernández Hernández, F Legorreta García, L E Hernández Cruz et al. This content was downloaded from IP address 170.106.35.234 on 24/09/2021 at 20:32 International Conference on Advanced Materials for Better Future 2016 IOP Publishing IOP Conf. Series: Materials Science and Engineering 176 (2017) 012039 doi:10.1088/1757-899X/176/1/012039 International Conference on Recent Trends in Physics 2016 (ICRTP2016) IOP Publishing Journal of Physics: Conference Series 755 (2016) 011001 doi:10.1088/1742-6596/755/1/011001 Impurities Removal in Seawater to Optimize the Magnesium Extraction N C Natasha1 , F Firdiyono1 and E Sulistiyono1 1Research Center of Metallurgy and Materials, LIPI, Puspiptek Gedung 470, Indonesia E-mail: [email protected] Abstract. Magnesium extraction from seawater is promising way because magnesium is the second abundant element in seawater and Indonesia has the second longest coastline in the world. To optimize the magnesium extraction, the impurities in seawater need to be eliminated. Evaporation and dissolving process were used in this research to remove the impurities especially calcium in seawater. -

Chemical Equilibrium 4
CHAPTER Chemical Equilibrium 4 ow we arrive at the point where real chemistry begins. Chemical thermo- Thermodynamic background dynamics is used to predict whether a mixture of reactants has a sponta- 4.1 The reaction Gibbs energy neous tendency to change into products, to predict the composition of the ⌬ N 4.2 The variation of rG with reaction mixture at equilibrium, and to predict how that composition will be mod- composition ified by changing the conditions. In biology, life is the avoidance of equilibrium, 4.3 Reactions at equilibrium and the attainment of equilibrium is death, but knowing whether equilibrium lies CASE STUDY 4.1: Binding of in favor of reactants or products under certain conditions is a good indication of oxygen to myoglobin and the feasibility of a biochemical reaction. Indeed, the material we cover in this chap- hemoglobin ter is of crucial importance for understanding the mechanisms of oxygen transport 4.4 The standard reaction in blood, metabolism, and all the processes going on inside organisms. Gibbs energy There is one word of warning that is essential to remember: thermodynamics is silent about the rates of reaction. All it can do is to identify whether a particular re- The response of equilibria to the conditions action mixture has a tendency to form products; it cannot say whether that ten- dency will ever be realized. We explore what determines the rates of chemical re- 4.5 The presence of a catalyst actions in Chapters 6 through 8. 4.6 The effect of temperature Coupled reactions in Thermodynamic background bioenergetics 4.7 The function of adenosine The thermodynamic criterion for spontaneous change at constant temperature and triphosphate pressure is ⌬G Ͻ 0. -
![Web of Science [V.5.28] - WOS Export Transfer Service](https://docslib.b-cdn.net/cover/1953/web-of-science-v-5-28-wos-export-transfer-service-9881953.webp)
Web of Science [V.5.28] - WOS Export Transfer Service
Web of Science [v.5.28] - WOS Export Transfer Service http://ets.webofknowledge.com/ETS/ets.do?SID=E3coQ5BgUvSvQ9dgbg6&product=... Close Web of Science Print Page 1 (Records 1 -- 50) [ 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 ] Record 1 of 491 Title: Raman antenna effect from exciton-phonon coupling in organic semiconducting nanobelts Author(s): Wang, M (Wang, Mao); Gong, Y (Gong, Yi); Alzina, F (Alzina, Francesc); Svoboda, O (Svoboda, Ondrej); Ballesteros, B (Ballesteros, Belen); Torres, CMS (Sotomayor Torres, Clivia M.); Xiao, SB (Xiao, Senbo); Zhang, ZL (Zhang, Zhiliang); He, JY (He, Jianying) Source: NANOSCALE Volume: 9 Issue: 48 Pages: 19328-19336 DOI: 10.1039/c7nr07212k Published: DEC 28 2017 Abstract: The highly anisotropic interactions in organic semiconductors together with the soft character of organic materials lead to strong coupling between nuclear vibrations and exciton dynamics, which potentially results in anomalous electrical, optical and optoelectrical properties. Here, we report on the Raman antenna effect from organic semiconducting nanobelts 6,13-dichloropentacene (DCP), resulting from the coupling of molecular excitons and intramolecular phonons. The highly ordered crystalline structure in DCP nanobelts enables the precise polarization-resolved spectroscopic measurement. The angle-dependent Raman spectroscopy under resonant excitation shows that all Raman modes from the skeletal vibrations of DCP molecule act like a nearly perfect dipole antenna I-Raman proportional to cos(4)(theta - 90), with almost zero (maximum) Raman scattering parallel (perpendicular) to the nanobelt's long-axis. The Raman antenna effect in DCP nanobelt is originated from the coupling between molecular skeletal vibrations and intramolecular exciton and the confinement of intermolecular excitons. -

Chemical Delivery Programme a Strong Connection for Quality and Safety STOCKMEIER Chemie
Chemical delivery programme A strong connection for quality and safety STOCKMEIER Chemie Customer proximity taken to the next stage We are more than just on site As a family business with over 90 years of experience, we have excellent connections to first-class producers. Through our customer relationships, we know the requirements for the most diverse applications. We always deliver in the right quantity, at the right market price, on time and to the desired location. Our product portfolio guarantees targeted solutions for different and cross-application requirements. And you have access to the comprehensive know-how of the entire STOCKMEIER Group at all times: STOCKMEIER Chemie + STAUB & CO. – KAPP-CHEMIE SILBERMANN + BASSERMANN minerals + Development and production of flexographic DE NOORD + HDS-Chemie printing inks and glues for the tissue industry, Dealing in standard chemicals and effect and process chemicals for the textile industry specialities, lime and other industrial as well as individual contract manufacturing. minerals. Development and manufacture STOCKMEIER Urethanes of cleaning agents for food-processing Development and production of polyurethane businesses / for surface technology / systems automotive / contract manufacturing STOCKMEIER Food KRUSE Automotive Development and production of flavourings System solution for AdBlue® and food ingredients as well as dealing in raw materials and additives for the food industry. 2 Customer proximity taken Contents to the next stage We are more than just on site About the product 04 Quality management 05 Solvents and softening agents 06 Acids and bases 10 Solids 12 Water treatment 16 Swimming pool chemicals 18 Services 20 Benefits at a glance 22 3 STOCKMEIER Chemie More than just products Our service is clear added value We develop, produce, and support our customers far beyond the traditional chemicals trade. -

Brno University of Technology Vysoké Učení Technické V Brně
BRNO UNIVERSITY OF TECHNOLOGY VYSOKÉ UČENÍ TECHNICKÉ V BRNĚ FACULTY OF CHEMISTRY FAKULTA CHEMICKÁ INSTITUTE OF CHEMISTRY AND TECHNOLOGY OF ENVIRONMENTAL PROTECTION ÚSTAV CHEMIE A TECHNOLOGIE OCHRANY ŽIVOTNÍHO PROSTŘEDÍ DEGRADATION OF HEAT TRANSFER FLUIDS IN THERMAL SOLAR SYSTEMS AND PROPANE-1,3-DIOL AS A NEW OPTION STÁRNUTÍ TEPLONOSNÝCH KAPALIN V TERMICKÝCH SOLÁRNÍCH SYSTÉMECH A PROPAN-1,3-DIOL JAKO NOVÁ MOŽNOST DOCTORAL THESIS DIZERTAČNÍ PRÁCE AUTHOR Ing. František Mikšík AUTOR PRÁCE SUPERVISOR prof. Ing. Josef Čáslavský, CSc. ŠKOLITEL BRNO 2018 Zadání dizertační práce Ústav: Ústav chemie a technologie ochrany životního prostředí Student: Ing. František Mikšík Studijní program: Chemie a technologie ochrany životního prostředí Studijní obor: Chemie životního prostředí Vedoucí práce: prof. Ing. Josef Čáslavský, CSc. Akademický rok: 2017/18 Název dizertační práce: Stárnutí teplonosných kapalin v termických solárních systémech a propan–1,3–diol jako nová možnost Zadání dizertační práce: 1. Dlouhodobé sledování a analýza provozu experimentálního termického solárního systému. 2. Identifikace procesů stárnutí teplonosných kapalin na bázi glykolů v termických solárních systémech, určení a charakterizace produktů degradace. 3. Analýza fyzikálních vlastností binární směsi propan–1,3–diolu s vodou. 4. Propan–1,3–diol jako moderní nemrznoucí teplonosná kapalina. Termín odevzdání dizertační práce: 8.6.2018 Ing. František Mikšík prof. Ing. Josef Čáslavský, CSc. doc. Ing. Jiří Kučerík, Ph.D. student(ka) vedoucí práce vedoucí ústavu prof. Ing. Martin Weiter, Ph.D. V Brně dne 1.9.2017 děkan Fakulta chemická, Vysoké učení technické v Brně / Purkyňova 464/118 / 612 00 / Brno ABSTRACT Degradation of heat transfer fluids on an organic basis is a long-standing problem known since the beginning of their use. -

Hydrazine Compound and Silver Halide Photographic Material Containing the Same
Europaisches Patentamt J European Patent Office © Publication number: 0 652 470 A1 Office europeen des brevets © EUROPEAN PATENT APPLICATION © Application number: 94117693.5 © Int. CI.6: G03C 1/06, C07D 257/04, C07D 487/04 @ Date of filing: 09.11.94 © Priority: 10.11.93 JP 303307/93 © Applicant: FUJI PHOTO FILM CO., LTD. 210 Nakanuma @ Date of publication of application: Minami-Ashigara-shi 10.05.95 Bulletin 95/19 Kanagawa 250-01 (JP) © Designated Contracting States: @ Inventor: Hioki, Takanori, c/o Fuji Photo Film BE DE FR GB IT NL Co.,Ltd. 210, Nakanuma Minami Ashigara-shi, Kanagawa (JP) Inventor: Ikeda, Tadashi, c/o Fuji Photo Film Co.,Ltd. 210, Nakanuma Minami Ashigara-shi, Kanagawa (JP) © Representative: Patentanwalte Grunecker, Kinkeldey, Stockmair & Partner Maximilianstrasse 58 D-80538 Munchen (DE) © Hydrazine compound and silver halide photographic material containing the same. © A silver halide photographic material comprising a support having thereon a silver halide emulsion layer which comprises a hydrazine compound having at least one absorbent group to silver halide, wherein two nitrogen atoms of the hydrazine compound are substituted by four substituents and the carbon atoms in the four substituents bonded directly to the two nitrogen atoms are not substituted by an oxo group. CM m CO Rank Xerox (UK) Business Services (3. 10/3.09/3.3.4) EP 0 652 470 A1 FIELD OF THE INVENTION The present invention relates to a silver halide photographic material having a high sensitivity and an excellent storage stability. 5 The present invention also relates to a novel compound. BACKGROUND OF THE INVENTION Silver halide photographic materials have hitherto been demanded to have a high sensitivity. -

Oxalate Oxalate (IUPAC: Ethanedioate) Is the Dianion with 2− 2− Oxalate the Formula C2O4 , Also Written (COO)2
Oxalate Oxalate (IUPAC: ethanedioate) is the dianion with 2− 2− Oxalate the formula C2O4 , also written (COO)2 . Either name is often used for derivatives, such as salts of oxalic acid, for example sodium oxalate Na2C2O4, or dimethyl oxalate ((CH3)2C2O4). Oxalate also forms coordination compounds where it is sometimes abbreviated as ox. Many metal ions form insoluble precipitates with oxalate, a prominent example being calcium oxalate, the primary constituent of the most common kind of kidney The structure of the oxalate anion stones. Contents Relationship to oxalic acid Structure Occurrence in nature Physiological effects A ball-and-stick model of oxalate As a ligand Names Excess IUPAC names [1][2] [3] Acquired oxalate , ethanedioate Congenital Systematic IUPAC name ethanedioate See also Other names References oxalate ion,[1] ethanedioic acid dianion, oxalic acid dianion[2] Relationship to oxalic acid Identifiers CAS Number 338-70-5 (http://www The dissociation of protons from oxalic acid proceeds in .commonchemistry.o a stepwise manner as for other polyprotic acids. Loss of rg/ChemicalDetail.as a single proton results in the monovalent px?ref=338-70-5) https://en.wikipedia.org/wiki/Oxalate 12/15/19, 12:49 PM Page 1 of 10 − [4][2] hydrogenoxalate anion HC2O4. A salt with this anion is sometimes called an acid oxalate, monobasic 3D model Interactive image (htt (JSmol) oxalate, or hydrogen oxalate. The equilibrium ps://chemapps.stolaf. −2 constant (Ka) for loss of the first proton is 5.37 × 10 edu/jmol/jmol.php?m (pKa = 1.27). The loss of the second proton, which yields odel=C%28%3DO% the oxalate ion, has an equilibrium constant of 29%28C%28%3DO −5 5.25 × 10 (pKa = 4.28).