Genetic Structure of SARS-Cov-2 in Western Germany Reflects Clonal Superspreading and Multiple Independent Introduction Events
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Konfirmationen 2021
Evangelische Kirchengemeinde Gangelt, Selfkant, Waldfeucht Gemeindebrief September/ Oktober/ November 2021 Konfirmationen 2021 1 Dürfen wir vorstellen: Konfirmation ist am Sonntag, 03.10.2021 im Geusen-Haus zu Bocket, An der Flachsroth 4 in 52525 Waldfeucht-Bocket Die Amtshandlungen werden nur in der Print-Ausgabe veröffentlicht 2 Foto: B. Hoppe Ein Wort zuvor Liebe Leserin, Foto: privat lieber Leser! Liebe Gemeinde, mit diesen Zeilen erreicht Sie die Herbstausgabe unseres Gemeindebriefs. Nach wie vor ist es eine verkürzte Ausgabe. Und doch enthält er das ganze Spektrum menschlichen Lebens. Wir sind angefüllt von den Katastrophenbildern vergangener Wochen. Auch Menschen und Gemeinden unseres Kirchenkreises wurde von den Hochwassern schwer getroffen. Die Corona-Lage bleibt unübersichtlich. Wenn auch Gottesdienste stattfinden können und auch die Unterrichte hoffentlich wieder präsentisch stattfinden werden, so ist die Zahl der Gemeindeveranstaltungen recht übersichtlich. Das liegt auch an unserem Bauvorhaben in Gangelt. Das Gemeindezentrum Friedenskirche ist nun erst einmal still gelegt. Allein das Gemeindebüro bleibt für Sie geöffnet. Die Redaktion hat sich entschieden, mit dem Stichwort „Konfirmationen 2021“ eine schöne Seite unseres Gemeindelebens in den Vordergrund zu rücken. Wir werden am 3. Oktober 14 junge Menschen in die Freiheit und Verantwortung des Glaubens entsenden und dieses in den Gottesdiensten feiern. Darauf dürfen wir uns gemeinsam freuen. Ihnen und Ihren Lieben Gesundheit, Lebensfreude und Gottes reichen Segen, Ihr Pfarrer Mathias Schoenen 3 Inhalt Unsere Konfirmanden/innen 2 Ein Wort zuvor 3 Andacht 5 Jubiläum Weltladen 6 Wir bauen ein Haus 7 Abschied v. K.-H. Palmen 8 Gemeindegruppen 9 Aus der Region 10 Brief des Präses 11 Aus dem Kirchenkreis 12 Adressen 14 Amtshandlungen 15 Gottesdienste 16 Impressum Gemeindebrief der Evangelischen Kirchengemeinde Gangelt, Selfkant, Waldfeucht Herausgeber: Presbyterium Redaktion: Lothar Kötz, Dr. -

Case Study North Rhine-Westphalia
Contract No. 2008.CE.16.0.AT.020 concerning the ex post evaluation of cohesion policy programmes 2000‐2006 co‐financed by the European Regional Development Fund (Objectives 1 and 2) Work Package 4 “Structural Change and Globalisation” CASE STUDY NORTH RHINE‐WESTPHALIA (DE) Prepared by Christian Hartmann (Joanneum Research) for: European Commission Directorate General Regional Policy Policy Development Evaluation Unit CSIL, Centre for Industrial Studies, Milan, Italy Joanneum Research, Graz, Austria Technopolis Group, Brussels, Belgium In association with Nordregio, the Nordic Centre for Spatial Development, Stockholm, Sweden KITE, Centre for Knowledge, Innovation, Technology and Enterprise, Newcastle, UK Case Study – North Rhine‐Westphalia (DE) Acronyms BERD Business Expenditure on R&D DPMA German Patent and Trade Mark Office ERDF European Regional Development Fund ESF European Social Fund EU European Union GERD Gross Domestic Expenditure on R&D GDP Gross Domestic Product GRP Gross Regional Product GVA Gross Value Added ICT Information and Communication Technology IWR Institute of the Renewable Energy Industry LDS State Office for Statistics and Data Processing NGO Non‐governmental Organisation NPO Non‐profit Organisation NRW North Rhine‐Westphalia NUTS Nomenclature of Territorial Units for Statistics PPS Purchasing Power Standard REN Rational Energy Use and Exploitation of Renewable Resources R&D Research and Development RTDI Research, Technological Development and Innovation SME Small and Medium Enterprise SPD Single Programming Document -

Interreg I / Ii : Cross-Border Cooperation
INTERREG I / II : CROSS-BORDER COOPERATION Euregio Meuse-Rhine: implementation and management in practice Speech by Mr K.H. Lambertz - Chair of the Monitoring Committee for the Euregio Meuse-Rhine Interreg Programme - Director of Euregio Meuse-Rhine - Minister-President of the German-speaking community of Belgium 1. General background and geographical situation In 1974, the governors of the Dutch and Belgian provinces of Limburg, together with the Chief Executive of the Cologne county administration, acted on the proposal made to them by the future Queen of the Netherlands, Princess Beatrix, during an official visit to Maastricht, to draw up draft arrangements for an association under which even closer cross-border collaboration could develop, along the lines of the Dutch-German Euregio project that had been running since 1957. This initiative was part of the new Community direction in regional policy, which in 1975 was to be provided with an instrument to assist economic development called the European Regional Development Fund (ERDF). In 1976, the principle of cross-border institutions was passed in law. Initially formed as an ad hoc association, the Euregio Meuse-Rhine was designed to promote integration between inhabitants on each side of the national borders. The area covers: • in Holland: the southern part of the Dutch province of Limburg; • in Germany: the city of Aachen, and the districts of Aachen, Heinsberg, Düren and Euskirchen, which make up the Aachen Regio, and • in Belgium: the entire province of Limburg. The province of Liège joined the Euregio Meuse-Rhine in 1978. In 1992, the German-speaking community of Belgium became the fifth partner in the Euregio Meuse- Rhine. -

The Districts of North Rhine-Westphalia
THE DISTRICTS OF NORTH RHINE-WESTPHALIA S D E E N R ’ E S G N IO E N IZ AL IT - G C CO TIN MPETENT - MEE Fair_AZ_210x297_4c_engl_RZ 13.07.2007 17:26 Uhr Seite 1 Sparkassen-Finanzgruppe 50 Million Customers in Germany Can’t Be Wrong. Modern financial services for everyone – everywhere. Reliable, long-term business relations with three quarters of all German businesses, not just fast profits. 200 years together with the people and the economy. Sparkasse Fair. Caring. Close at Hand. Sparkassen. Good for People. Good for Europe. S 3 CONTENTS THE DIstRIct – THE UNKnoWN QUAntITY 4 WHAT DO THE DIstRIcts DO WITH THE MoneY? 6 YoUTH WELFARE, socIAL WELFARE, HEALTH 7 SecURITY AND ORDER 10 BUILDING AND TRAnsPORT 12 ConsUMER PRotectION 14 BUSIness AND EDUCATIon 16 NATURE conseRVAncY AND enVIRonMentAL PRotectIon 18 FULL OF LIFE AND CULTURE 20 THE DRIVING FORce OF THE REGIon 22 THE AssocIATIon OF DIstRIcts 24 DISTRIct POLICY AND CIVIC PARTICIPATIon 26 THE DIRect LIne to YOUR DIstRIct AUTHORITY 28 Imprint: Editor: Dr. Martin Klein Editorial Management: Boris Zaffarana Editorial Staff: Renate Fremerey, Ulrich Hollwitz, Harald Vieten, Kirsten Weßling Translation: Michael Trendall, Intermundos Übersetzungsdienst, Bochum Layout: Martin Gülpen, Minkenberg Medien, Heinsberg Print: Knipping Druckerei und Verlag, Düsseldorf Photographs: Kreis Aachen, Kreis Borken, Kreis Coesfeld, Ennepe-Ruhr-Kreis, Kreis Gütersloh, Kreis Heinsberg, Hochsauerlandkreis, Kreis Höxter, Kreis Kleve, Kreis Lippe, Kreis Minden-Lübbecke, Rhein-Kreis Neuss, Kreis Olpe, Rhein-Erft-Kreis, Rhein-Sieg-Kreis, Kreis Siegen-Wittgenstein, Kreis Steinfurt, Kreis Warendorf, Kreis Wesel, project photos. © 2007, Landkreistag Nordrhein-Westfalen (The Association of Districts of North Rhine-Westphalia), Düsseldorf 4 THE DIstRIct – THE UNKnoWN QUAntITY District identification has very little meaning for many people in North Rhine-Westphalia. -

The Germans of Roberts Cove, Louisiana: German Rice Cultivation and the Making of a German-American Community in Acadia Parish, 1881-1917
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 12-17-2010 The Germans of Roberts Cove, Louisiana: German Rice Cultivation and the Making of a German-American Community in Acadia Parish, 1881-1917 Lydia Soileau University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Soileau, Lydia, "The Germans of Roberts Cove, Louisiana: German Rice Cultivation and the Making of a German-American Community in Acadia Parish, 1881-1917" (2010). University of New Orleans Theses and Dissertations. 1246. https://scholarworks.uno.edu/td/1246 This Thesis is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Thesis in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights- holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/or on the work itself. This Thesis has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. The Germans of Roberts Cove, Louisiana: German Rice Cultivation and the Making of a German-American Community in Acadia Parish, 1881-1917 A Thesis Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the requirements for the degree of Master of Arts In History By Lydia Alicia Soileau B.A. -

I Online Supplementary Data – Lötters, S. Et Al.: the Amphibian Pathogen Batrachochytrium Salamandrivorans in the Hotspot Of
Online Supplementary data – Lötters, S. et al.: The amphibian pathogen Batrachochytrium salamandrivorans in the hotspot of its European invasive range: past – present – future. – Salamandra, 56: 173–188 Supplementary document 1. Published site records (populations) of caudate species from Germany in which Bsal was detected until 2018. Data mostly summarized from Spitzen-van der Sluijs et al. (2016), Dalbeck et al. (2018), Lötters et al. (2018), Schulz et al. (2018) and Wagner et al. (2019a). In addition, new findings from the ongoing laboratory testing (especially quality assurance) of samples collected in same time frame were also included, so that some entries differ from those in the mentioned articles. Specimens tested positive for Bd/Bsal and negative for only Bd are indicated under remarks. Legend: † = dead specimen(s); + = ‘low’ infection load (1–10 GE); ++ = ‘medium’ infection load (> 10–100 GE); +++ = ‘high’ infection load (> 100 GE); CI = credible interval per year. Site District Coordinates Species Year N samples N samples Infection Prevalence 95% Remarks (latitude, tested Bsal- loads per year Bayesian longitude) positive CI Northern Eifel North Rhine-Westphalia, StädteRegion 50.578169, Fire salamander, Salamandra salamandra 2015 22 (of which 21 (of which 96% 79–99% mass mortality, 8 of 16 specimens Belgenbach Aachen 6.278448 16 †) 16 †) had Bd/Bsal co-infections Fire salamander, Salamandra salamandra 2017 12 larvae 0 0 0% 0–26% North Rhine-Westphalia, StädteRegion 50.746724, Northern crested newt, Triturus cristatus 2015 2 -

Europa Regina. 16Th Century Maps of Europe in the Form of a Queen Europa Regina
Belgeo Revue belge de géographie 3-4 | 2008 Formatting Europe – Mapping a Continent Europa Regina. 16th century maps of Europe in the form of a queen Europa Regina. Cartes d’Europe du XVIe siècle en forme de reine Peter Meurer Electronic version URL: http://journals.openedition.org/belgeo/7711 DOI: 10.4000/belgeo.7711 ISSN: 2294-9135 Publisher: National Committee of Geography of Belgium, Société Royale Belge de Géographie Printed version Date of publication: 31 December 2008 Number of pages: 355-370 ISSN: 1377-2368 Electronic reference Peter Meurer, “Europa Regina. 16th century maps of Europe in the form of a queen”, Belgeo [Online], 3-4 | 2008, Online since 22 May 2013, connection on 05 February 2021. URL: http:// journals.openedition.org/belgeo/7711 ; DOI: https://doi.org/10.4000/belgeo.7711 This text was automatically generated on 5 February 2021. Belgeo est mis à disposition selon les termes de la licence Creative Commons Attribution 4.0 International. Europa Regina. 16th century maps of Europe in the form of a queen 1 Europa Regina. 16th century maps of Europe in the form of a queen Europa Regina. Cartes d’Europe du XVIe siècle en forme de reine Peter Meurer 1 The most common version of the antique myth around the female figure Europa is that which is told in book II of the Metamorphoses (“Transformations”, written around 8 BC) by the Roman poet Ovid : Europa was a Phoenician princess who was abducted by the enamoured Zeus in the form of a white bull and carried away to Crete, where she became the first queen of that island and the mother of the legendary king Minos. -

2 Years 255 Days
Volume 29, No. 9 NATO Air Base Geilenkirchen 27 Septembery 2013 Photo by Wiel Borghans Exercise, Exercise, Exercise The E-3A Component held a large, multi-scenario exercise in cooridination with local German emergency personnel. For more photos and information, see pages 8 and 9. Training Wing changes command Colonel Arturo Di Martino assumes command of the Training Wing on Sept. 20, 2013. Photo by Wiel Borghans By Maureen Geraets-Head The new Training Wing Commander, Colonel Di Martino said in his The Trapani Room at the E-3A Club speech, “I am fully aware of the huge is filled with guests attending the responsibilities that are connected with Training Wing Change of Command. the fulfillment of a Wing Commander’s duties. Being in charge as the Wing With the marching in of the Colors Commander certainly means to be the ceremony starts. Colonel Arturo accountable. Accountable, not only to Di Martino, ITAAF,60¡ officially65¡ assumes70¡ the Component75¡ 80¡ Commander,85¡ not only90¡ 95¡ 100¡ 105¡ 110¡ 115¡ 120¡ 125¡ 40¡ command of theUZBEKIS TrainingTAN Wing on to the flag that I’m representing in this KAZAKHST AN Sept. 20, 2013, after that Lt. Col. multinational environment,Dzunga but alsor ia to MONGOLIA Almaty Knud Holmsgaard has relinquished theBishkek other Wing Commanders†rŸmqi by giving GOBI Beijing Dalian Bukhara Oz. Issyk Kul' N command. Tashkent them the support and outputH thatA UZBEKISTAN KYRGYZSTAN S Hami Baotou Tianjin Bo Hai N Samarkand E they are expectingI from one’s unitT urinfan a Depression Aksu TURKMENISTAN T 35¡ Maj.Mashhad Gen. Andrew Mueller presided timely, proper manner, always keeping ORDOS Shijiazhuang 35¡ Dushanbe Qingdao over the change of command. -

Ansprechpartnerinnen Und Ansprechpartner
Ansprechpartnerinnen und Ansprechpartner 1 Amtshaftung ............................................................................................................... 4 1.1 Sachbearbeitung Amtshaftung ............................................................................. 4 1.2 Teildezernatsleitung ............................................................................................. 4 2 Dienstunfall ................................................................................................................. 4 2.1 Sachbearbeitung Dienstunfall ............................................................................... 4 2.2 Teildezernatsleitung ............................................................................................. 5 3 Einstellung/ Versetzung .............................................................................................. 5 3.1 Berufskolleg / Seiteneinstieg ................................................................................ 5 3.2 Förderschule ........................................................................................................ 6 3.3 Gesamtschule ...................................................................................................... 6 3.4 Grundschule ......................................................................................................... 6 3.5 Hauptschule/Sekundarschule/ Gemeinschaftsschule/PRIMUS-Schule ................ 7 3.6 Gymnasium/Weiterbildungskolleg ....................................................................... -

A Historical and Linguistic Study of the German Settlement at Roberts Cove, Louisiana
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1969 A Historical and Linguistic Study of the German Settlement at Roberts Cove, Louisiana. Stanley Joe Mccord Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Mccord, Stanley Joe, "A Historical and Linguistic Study of the German Settlement at Roberts Cove, Louisiana." (1969). LSU Historical Dissertations and Theses. 1606. https://digitalcommons.lsu.edu/gradschool_disstheses/1606 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. This dissertation has been microfilmed exactly as received 70-253 McCORD, Stanley Joe, 1936- A HISTORICAL AND LINGUISTIC STUDY OF THE GERMAN SETTLEMENT AT ROBERTS COVE, LOUISIANA. [Portions of Text in German]. The Louisiana State University and Agricultural and Mechanical College, Ph.D., 1969 Language and Literature, modem University Microfilms, Inc., Ann Arbor, Michigan Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. A HISTORICAL AND LINGUISTIC STUDY OF THE GERMAN SETTLEMENT AT ROBERTS COVE, LOUISIANA A Dissertation Submitted, to the Graduate Faculty of the Louisiana State University and Agricultural and Mechanical College in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Foreign Languages fcy Stanley Joe McCord B,A.f Louisiana State University, i960 M.A., Louisiana State University, 1963 May, 1969 Reproduced with permission of the copyright owner. -

A Beacon for the COVID-19 Epidemic Control in Brasil: Seroepidemiological Population-Based Surveys Maria Rita Donalisio1*
LETTER TO THE EDITOR https://doi.org/10.1590/1806-9282.67.01.20200512 A beacon for the COVID-19 epidemic control in Brasil: seroepidemiological population-based surveys Maria Rita Donalisio1* The novel severe acute respiratory syndrome coronavirus 2 infection in the population are useful for assessing the impact (SARS-CoV-2), which was firstly identified in late December of public health interventions and can be used in clinical vac- 2019 in China, has spread rapidly worldwide. However, epi- cine trials. demics are expressed from different forms around the world. Population-based surveys have guided public health mea- The epidemic curves do not follow a “natural path” due to dif- sures in several countries. Cross-sectional studies in Spain have ferent social, epidemiological, and cultural contexts, access to shown the most affected regions around Madrid and the dis- health services and to diagnostic tests, vaccine, and preventive ease transmission9. Furthermore, various authors in Iceland measures in different regions1. Oliveira et al.2 investigated dis- have identified populations at risk10. A study conducted in parities in the clinical profile of coronavirus disease (COVID- Gangelt, Heinsberg district, in Germany, where the first case 19) and in the groups that can be more affected by the dis- of COVID-19 was announced in the country, reported that ease in Brasil, China and Italy since the pandemic beginning. 15% of the city population presented antibodies against SARS- In Brasil, the authors reported the highest percentage of cases CoV-2. The first surveys carried out in the United States, in among individuals aged <60, male, and diabetes-affected ones2. -
200 Dpi Resolution
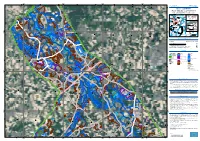
295000 300000 305000 310000 6°4'0"E 6°6'0"E 6°8'0"E 6°10'0"E 6°12'0"E 6°14'0"E 6°16'0"E 6°18'0"E GLIDE number: N/A Activation ID: EMSR517 N " Int. Charter Act. ID: N/A Product N.: 05BONN, v1 0 ' 8 ° 1 5 Bonn - GERMANY Flood - Situation as of 16/07/2021 Delineation MONIT01 - Detail map 04 Sachsen-Anhalt Nordrhein-Westfalen Niedersachsen Zuid-Nederland 06 Vlaams Gewest Detail 04 Detail 02 Detail 03 ThurinBagltice Snea (! Bonn 05 North Sea Region 02 Hessen Wallonne ^ Poland 03 Berlin Rheinland-Pfalz Germany Belgium Czech 01 Republic N France " Saarland 0 Austria ' 0 0 6 0 0 ° 1 Bayern 0 0 Italy 5 Switzerland 5 5 6 6 6 Wassenberg 6 ! 5 5 Est 60 Baden-Wurttemberg km N " 0 ' 6 ° 1 5 Cartographic Information 1:30000 Full color A1, 200 dpi resolution 0 0.5 1 2 km Grid: WGS 1984 UTM Zone 32N map coordinate system Tick marks: WGS 84 geographical coordinate system ± Legend Crisis Information Built-Up Area Hydrography Flooded Area Residential River (16/07/2021 05:41 UTC) General Information Office Stream Area of Interest Wholesale and retail trade Lake Administrative Industrial Land Subject to Inundation boundaries N School, university and research " Reservoir 0 ' International Boundary 4 ° 1 5 River Province Hospital or institutional care Heinsberg Facilities ! Placenames Military ! Dam Placename Cemetery Transportation N Highway Nordrhein-Westfalen " 0 ' 4 ° 1 Primary Road 5 Secondary Road Land Use - Land Cover 0 0 Features available in the vector package 0 0 0 0 0 0 6 6 6 6 5 5 Hückelhoven ! Map Information Heavy rains affected Rhineland-Palatinate area where a severe flood event is expected over E " 0 ' the next few days along the river Moselle.