Interfacial Adhesion in Rayon/Nylon Sheath/Core Composite Fibers. Weiying Tao Louisiana State University and Agricultural & Mechanical College
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
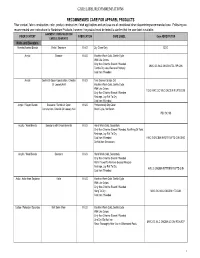
Care Label Recommendations
CARE LABEL RECOMMENDATIONS RECOMMENDED CARE FOR APPAREL PRODUCTS Fiber content, fabric construction, color, product construction, finish applications and end use are all considered when determining recommended care. Following are recommended care instructions for Nordstrom Products, however; the product must be tested to confirm that the care label is suitable. GARMENT/ CONSTRUCTION/ FIBER CONTENT FABRICATION CARE LABEL Care ABREVIATION EMBELLISHMENTS Knits and Sweaters Acetate/Acetate Blends Knits / Sweaters K & S Dry Clean Only DCO Acrylic Sweater K & S Machine Wash Cold, Gentle Cycle With Like Colors Only Non-Chlorine Bleach If Needed MWC GC WLC ONCBIN TDL RP CIIN Tumble Dry Low, Remove Promptly Cool Iron If Needed Acrylic Gentle Or Open Construction, Chenille K & S Turn Garment Inside Out Or Loosely Knit Machine Wash Cold, Gentle Cycle With Like Colors TGIO MWC GC WLC ONCBIN R LFTD CIIN Only Non-Chlorine Bleach If Needed Reshape, Lay Flat To Dry Cool Iron If Needed Acrylic / Rayon Blends Sweaters / Gentle Or Open K & S Professionally Dry Clean Construction, Chenille Or Loosely Knit Short Cycle, No Steam PDC SC NS Acrylic / Wool Blends Sweaters with Embelishments K & S Hand Wash Cold, Separately Only Non-Chlorine Bleach If Needed, No Wring Or Twist Reshape, Lay Flat To Dry Cool Iron If Needed HWC S ONCBIN NWOT R LFTD CIIN DNID Do Not Iron Decoration Acrylic / Wool Blends Sweaters K & S Hand Wash Cold, Separately Only Non-Chlorine Bleach If Needed Roll In Towel To Remove Excess Moisture Reshape, Lay Flat To Dry HWC S ONCBIN RITTREM -

Choosing the Proper Short Cut Fiber for Your Nonwoven Web
Choosing The Proper Short Cut Fiber for Your Nonwoven Web ABSTRACT You have decided that your web needs a synthetic fiber. There are three important factors that have to be considered: generic type, diameter, and length. In order to make the right choice, it is important to know the chemical and physical characteristics of the numerous man-made fibers, and to understand what is meant by terms such as denier and denier per filament (dpf). PROPERTIES Denier Denier is a property that varies depending on the fiber type. It is defined as the weight in grams of 9,000 meters of fiber. The current standard of denier is 0.05 grams per 450 meters. Yarn is usually made up of numerous filaments. The denier of the yarn divided by its number of filaments is the denier per filament (dpf). Thus, denier per filament is a method of expressing the diameter of a fiber. Obviously, the smaller the denier per filament, the more filaments there are in the yarn. If a fairly closed, tight web is desired, then lower dpf fibers (1.5 or 3.0) are preferred. On the other hand, if high porosity is desired in the web, a larger dpf fiber - perhaps 6.0 or 12.0 - should be chosen. Here are the formulas for converting denier into microns, mils, or decitex: Diameter in microns = 11.89 x (denier / density in grams per milliliter)½ Diameter in mils = diameter in microns x .03937 Decitex = denier x 1.1 The following chart may be helpful. Our stock fibers are listed along with their density and the diameter in denier, micron, mils, and decitex for each: Diameter Generic Type -

Investigation of the Mechanical Properties of a Carbon Fibre-Reinforced Nylon Filament for 3D Printing
machines Article Investigation of the Mechanical Properties of a Carbon Fibre-Reinforced Nylon Filament for 3D Printing Flaviana Calignano 1,* , Massimo Lorusso 2 , Ignanio Roppolo 3 and Paolo Minetola 1 1 Department of Management and Production Engineering, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy; [email protected] 2 Istituto Italiano di Tecnologia, Center for Sustainable Future Technologies IIT@Polito, Corso Trento 21, 10129 Turin, Italy; [email protected] 3 Department of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Turin, Italy; [email protected] * Correspondence: fl[email protected]; Tel.: +39-011-090-7218 Received: 19 July 2020; Accepted: 2 September 2020; Published: 4 September 2020 Abstract: Additive manufacturing (i.e., 3D printing) has rapidly developed in recent years. In the recent past, many researchers have highlighted the development of in-house filaments for fused filament fabrication (FFF), which can extend the corresponding field of application. Due to the limited mechanical properties and deficient functionality of printed polymer parts, there is a need to develop printable polymer composites that exhibit high performance. This study analyses the actual mechanical characteristics of parts fabricated with a low-cost printer from a carbon fibre-reinforced nylon filament. The results show that the obtained values differ considerably from the values presented in the datasheets of various filament suppliers. Moreover, the hardness and tensile strength are influenced by the building direction, the infill percentage, and the thermal stresses, whereas the resilience is affected only by the building direction. Furthermore, the relationship between the mechanical properties and the filling factor is not linear. -

Nylon Wool Fiber Columns
U.S. Corporate Headquarters Polysciences Europe GmbH Polysciences Asia-Pacific, Inc. 400 Valley Rd. Badener Str. 13 2F-1, 207 DunHua N. Rd. Warrington, PA 18976 69493 Hirschberg an der Taipei, Taiwan 10595 1(800) 523-2575 / (215) 343-6484 Bergstrasse, Germany (886) 2 8712 0600 1(800)343-3291 fax +(49) 6201 845 20 0 (886) 2 8712 2677 fax [email protected] +(49) 6201 845 20 20 fax [email protected] [email protected] TECHNICAL DATA SHEET 425A Page 1 of 2 Nylon Wool Fiber Columns BACKGROUND NYLON WOOL FIBER VS. SHEEP RBC ROSETTING METHODS Researchers have been using nylon wool fiber procedures to separate T-cell and Wong and Mittal (1981)9 did extensive research comparing the methods of Nylon B-cell lymphocytes for more than 20 years. In the early 1970’s M. H. Julius et al Wool Fiber separation and the commonly- used and well-studied sheep RBC (1973),1 Eisen et al (1972),2 and Greaves & Brain (1974)3 described specific (SRBC) rosetting.10,11 Wong and Mittal were interested in isolating B-cells for conditions for the use of Nylon Wool Fiber in columns or plastic straws. These serologic typing of HLA-DR antigen. protocols resulted in yields of 50-90% T-cell recovery and 10-100 fold B-cell depletion. Wong and Mittal concluded that “Due to its simplicity and reliability, nylon wool adherence may be preferred over the SRBC rosette method for the routine pheno- These early researchers found it necessary to scrub or wash their Nylon Wool Fiber typing of B-cells.” Their findings are illustrated in Table 1. -

Immersion Dyeing Nylon and Acetate Rayon Using Prosperse Disperse Dyes Please Read Directions Carefully Before Starting
Immersion Dyeing Nylon and Acetate Rayon using PROsperse Disperse Dyes Please read directions carefully before starting. For medium to dark shades, it is recommended that nylon be dyed with acid dyes, because disperse dyes lack acceptable fastness. Acetate rayon can only be dyed with disperse dyes and has acceptable fastness in all depths of shade with the disperse dyes. All Dyeing should be done in a stainless steel or enamelware container only. Never use aluminum pots. Use Pyrex or stainless steel measuring utensils and a large wooden dowel for stirring in the boiling dye bath. Always do test samples before working on a large project. Please Note: These dyes have the potential to stain any sink that is not made of stainless steel or fireclay ceramic. For additional information, visit our web site at www.prochemicalanddye.com. Wear rubber gloves, apron, or old clothes. Utensils used for dyeing should never be used for food preparation. Supplies PROsperse Disperse Dye Citric Acid Crystals or White Distilled Vinegar Synthrapol PRO Dye Activator or Soda Ash Procedure 1. Scour the fabric by machine washing in HOT 140F (60C) water, or by hand in a pot on the stove with 2 tsp (2 gm) PRO Dye Activator or Soda Ash and 2 tsp (2.5 ml) Synthrapol per pound of fabric (454 gm, or 3 to 4 yards of muslin weight fabric). Rinse thoroughly. This step does not add the dye fixative to the fabric; it prepares your fabric for dyeing by removing any dirt, oil or sizing. 2. Dissolve the dye. Thoroughly dissolve the desired amount of dye powder, from the chart below, in 1 cup (250 ml) of boiling water. -

Colaris Digital Textile Printing
ZIMMER AUSTRIA | DIGITAL PRINTING SYSTEMS COLARIS DIGITAL TEXTILE PRINTING HOME TEXTILES APPAREL DECORATION AUTOMOTIVE FLAGS & BANNERS www.zimmer-austria.com 2020.01.15 page 1 CONTENT 1. INNOVATION IS IN OUR DNA 1.1. HISTORIC MILESTONES 3 2. INK CLASSES 2.1. TYPES | PRODUCTS | PROCESS | REQUIREMENTS 4 2.2. TYPES | PRODUCTS | PROCESS | REQUIREMENTS 5 3. PRINT TECHNOLOGY 3.1. PROCESSING DIAGRAM 6 3.2. PROCESS EQUIPMENT 7 4. REACTIVE PRINTING 4.1. GENERAL INFORMATION 8 4.2. EXAMPLE: TERRY TOWEL PRINT PRODUCTION 9 5. ACID PRINTING 5.1. GENERAL INFORMATION 10 5.2. EXAMPLE: UPHOLSTERY PRINT LINE 11 6. DISPERSE / SUBLIMATION PRINTING 6.1. GENERAL INFORMATION 12 6.2. EXAMPLE: PES BLANKET PRINT LINE 13 7. VAT INDANTHRENE® PRINTING 7.1. GENERAL INFORMATION 14 7.2. APPLICATION DIVERSITY 15 8. PIGMENT PRINTING 8.1. GENERAL INFORMATION 16 8.2. APPLICATION DIVERSITY 17 9. CATIONIC PRINTING 9.1. GENERAL INFORMATION 18 10. COLARIS - CHARACTERISTICS AND FEATURES 10.1. COLARIS MODELS 19 11. COLARIS FEATURES AND COMPONENTS 11.1. INTEGRATED MACHINE COMPONENTS 20 11.2. INTEGRATED MACHINE COMPONENTS 21 12. PROCESS EQUIPMENT 12.1. INLINE COMPONENTS 22 12.2. OFFLINE COMPONENTS 23 13. PRINT HEAD 13.1. TECHNOLOGY 24 13.2. RECONDITION CENTER 25 14. ZIMMER TECHNOLOGY & APPLICATION CENTER 14.1. GENERAL INFORMATION 26 14.2. EQUIPMENT & FACILITIES 27 www.zimmer-austria.com 2020.01.15 page 2 1. INNOVATION IS IN OUR DNA 1.1. HISTORIC MILESTONES Vertical Duplex blanket printer from 1951 First commercial rotary screen printer 1958 The broad digital competence of ZIMMER AUSTRIA is based on an innovation introduced more than 4 decades ago. -

Fabric Fiber Content
Fabric Types, Count & Fiber Content Zweigart Linen Count Content Belfast 32 100% linen Afghans - 100% Polyacrylic Cashel 28 100% linen Abby 18ct Alba 14ct Almanac 14ct Cork 19 100% linen Anne Cloth 18ct Baby Snuggle 18ct Country Home 18ct Dublin 25 100% linen Diamond 18ct Gloria 14ct Hearthside 14ct Edinborough 36 100% linen Honeycomb 18ct Novara 14ct Patrice 14ct Fine Linen 45 55% linen + 45% cotton Afghans - 100% Cotton Glasgow 28 100% linen Anne Cloth 18ct Augusta 14 ct Novara 14ct Kingston 50 100% linen Teresa 14ct Newcastle 40 100% linen Afghans- Misc Normandie 55% cotton + 45% linen Pastel LinenD 28 52% cotton + 48% linen Gloria 14ct 70% rayon + 30% linen Pearl Linen 20, 25, 28 60% polyester + 40% linen Merino 28ct 100% Wool Mosaik 18ct 52% cotton + 42% rayon Patterned Count Content Tannenbaum 18ct 52% cotton + 42% rayon Cottage Huck 14 100% cotton Aida Weave Count Content Belinda 20 52% cotton + 48% rayon Diana 20 52% cotton + 48% rayon Aida 8, 11, 14, 16, 18 100% cotton Newport 28 100% linen Country AidaD 7 100% polyacrylic Sambuca 28 60% polyester + 40% linen Damask Aida 11,14,18 52% cotton + 48% rayon Saronno 28 52% cotton + 48% rayon GoldauD 7 55% rayon + Shenandoah 28 55% linen + 45% rayon 40% cotton + 5% metallic Hardanger 22 100% cotton Canvas Count Content Hearthstone 14 60% cotton + 40% linen Congress 24 100% cotton Herta 6 100% cotton Congressa 24 100% cotton Huck 14 100% cotton Cordova 22 100% cotton Klostern 7 60% rayon + 40% cotton Double Mesh 5, 6.5, 7.5, 10, 12, Linen Hardanger 22 100% linen 14, 16, 18, 20 100% cotton -

Evaluation of the Physical and Diagnostic Nature of Swabs W
EVALUATION OF THE PHYSICAL AND DIAGNOSTIC NATURE OF SWABS W. Nugent, M.D.1, M.J. Russell1 S. Beck 2, and G.P. Leonardi, Ph.D.1,3 NASSAU 1 M44 Department of Pathology, Nassau University Medical Center, East Meadow, NY ; UNIVERSITY Department of Biology, Nassau Community College, Garden City, NY2 and Sunrise Medical Laboratories, Hauppauge, NY3 medicalcenter Abstract: Conclusions: Proper specimen collection is the single most important factor in obtaining a reliable and and reweighed. Following addition of water (1.0ml), tubes were shaken at high speeds ß Fiber confi guration and size of the nylon-fl ocked swab allowed for greater accurate diagnostic result. Swabs are often employed to collect specimens for a variety for 1 minute, followed by centrifugation using a table-top Hermle Z180M centrifuge absorbance than the actual nylon material itself. of microbiologic assays. A novel, nylon-tipped, “fl ocked“ swab (Copan, Inc; Corona, CA) (Universal Labnet Co., Edison, NJ) for 5 minutes at maximum speed. Unabsorbed water FIG. 1 FIG. 2 has recently become commercially available. Unlike conventional swabs in which fi bers ß There are numerous other factors, not presently examined, which can aff ect was removed by decanting and wiping the inner tube surface with bibulous paper, swab performance. are woven on a shaft material, fl ocked swabs have a “one piece” construction with nylon and the tube was reweighed. This exercise was repeated twice. fi bers radiating from the shaft tip. This construction may enhance the amount of specimen ß Nevertheless, our data suggests that using of nylon-fl ocked swabs would available for diagnostic testing. -

Digital Textile Printing Has Come of Age
diversification Digital Textile Printing Has Come of Age One only needs to look at what is happening in other parts of the world to see that now is the time for companies in North America to invest resources to participate in the projected growth of digital textile printing. Digital textile printing has officially Italy, Turkey and Spain, along with Brazil entered a new age. e global market for and India, have embraced digital textile volume of printed textile is around 27 printing to the point where they have idled billion yards per year, growing at three some of their screen printing equipment in percent. It is estimated that about 250 favor of printing textiles digitally. ey are million yards is currently printed digitally. producing yardage of all types of products e growth curve of digitally printed yards including apparel, home furnishing, has steepened over the past two to three interior decoration and signage. years and it is expected that by 2016, the The major drawbacks of printing volume will be well over one billion yards. digitally, namely speed and cost have been, Does digital textile printing finally and continue to be, addressed. Digital make sense as a mainstream solution to printers now in production reach speeds serve the needs of the signage and other of 75 yards per minute. Companies are markets? Has the technology progressed realizing running costs in the range of to the point of it being viable to more than $0.50/yard compared to $0.35/yard for a few companies that have mastered its rotary screen printing. -

Silk and Rayon in Popular US Publications of the Nineteen Thirties
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Textile Society of America Symposium Proceedings Textile Society of America 2002 Competing Images: Silk and Rayon in Popular U.S. Publications of the Nineteen Thirties Jocelyn Gottschalk Fashion Institute of Technology Follow this and additional works at: https://digitalcommons.unl.edu/tsaconf Part of the Art and Design Commons Gottschalk, Jocelyn, "Competing Images: Silk and Rayon in Popular U.S. Publications of the Nineteen Thirties" (2002). Textile Society of America Symposium Proceedings. 520. https://digitalcommons.unl.edu/tsaconf/520 This Article is brought to you for free and open access by the Textile Society of America at DigitalCommons@University of Nebraska - Lincoln. It has been accepted for inclusion in Textile Society of America Symposium Proceedings by an authorized administrator of DigitalCommons@University of Nebraska - Lincoln. ( Competing Images: Silk and Rayon in Popular U.S. Publications of the Nineteen Thirties f Jocelyn Gottschalk / In 1934, Mr. Paolino Gerli, President of the newly formed International Silk Guild, concluded his address at the Twenty-third Annual Convention of the National Retail Dry Goods Association as follows: "A new deal, a new era. Out of a glamorous past, not a Cinderella with a time limit beauty is beckoning to you, but a beautiful Princess, awakened ( by Prince Charming into a new consciousness of its eternal right to be the Queen of Fabrics."1 These lofty words, delivered before an audience of American manufacturers, retailers, and wholesalers, were a call to arms for the beleaguered American silk industry. His words imparting a tone of righteousness, Mr. -

Fabric Supplier List
FABRIC SUPPLIER LIST CANADA Kendor Textiles Ltd 1260 Cliveden Ave Delta BC V3M 6Y1 Canada 604.434.3233 [email protected] www.kendortextiles.com Fabrics Available: Fabric supplier. Eco-friendly. Organic. Knits: solids, prints, yarn dyes and warp. Wovens: solids and yarn dyes. End Use: activewear, bottomweights, medical, lingerie, childrenswear, swimwear, rainwear, skiwear and uniform. Natural & eco items include cottons, bamboo's, modals, linens, hemps, organic cottons & organic linens. Technical items include waterproof/breathable soft shells, antibacteric & wicking polyester & recycled polyesters. Is a proud representative of the British Millerain line of waxed cottons and wools, and are able to provide custom souring. Minimums: Carries stock. In-stock minimum: 5 yards/color. Minimum order for production: 10 yards/color. Gordon Fabrics LTD #1135-6900 Graybar Rd. Richmond BC Canada 604.275.2672 [email protected] Fabrics Available: Fabric Supplier. Importer. Jobber. Carries stock. Knits & Wovens: solids, prints, yarn dyes and novelties. End Use: activewear, borromweights, eveningwear/bridal, medical, lingerie and childrenswear. Minimums: In stock minimum 1 yard. Minimum order for production varies. StartUp Fashion Supplier List 2016 – Page 1 CHINA Ecopel (HX) Co., Ltd. China +86 216.767.9686 www.ecopel.cn Fabrics Available: Fake fur and leather garments. End Uses: Childrenswear, Menswear, Other, Womenswear. Minimums: Min. order 50-100 m Hangzhou New Design Source Textile Co., Ltd. China +86 057.182.530528 Fabrics Available: Knits, Polyester/Man-Made, Prints. End Uses: Juniors Fashion, Menswear, Womenswear. Minimums: Min order 50 m. Nantong Haukai Textile Co., Ltd. China +86 513.890.78626 www.huakaitex.com Fabrics Available: Cotton, Linen. End Uses: Corporatewear/Suiting, Menswear, Womenswear. -

CLASSIC RAYON 100% Viscose
CLASSIC RAYON 100% viscose While every attempt is made to reproduce thread colors accurately, colors on your monitor or print out may not precisely match thread colors. Color names are for your reference only. When ordering please refer to each color by color number. 1013 Peach Blush 1111 Evening Mist 1015 Desert Bloom 1031 Frosted Lavender 1317 Blush Pink 1235 Crocus 1220 Conch Shell 1320 Purple Heart 1307 Raspberry Punch 1388 Plum 1485 Electric Red 1319 Iris 1039 Brick Red 1488 Dark Magenta 1038 Barn Red 1310 Magenta 1114 Pink Petal 1321 Bubble Gum Pink 1115 Powder Puff 1121 Candy Heart 1315 Pink Grapefruit 1309 Dahlia 1148 Rustic Pink 1109 Pink Rose 1384 Merlot 1110 Fuchsia 1385 Garnet 1383 Pink Pansy 1182 Mulberry 1187 Orchid 1281 Radish 1234 Hibiscus 1184 Scarlet Rose 1117 Flamingo Pink 1154 Lipstick Rose 1183 Cranberry 1107 Honeysuckle 1389 Bordeaux 1014 Bermuda Sand 1034 Vintage Rose 1120 Baby Pink 1119 English Rose 1116 Cotton Candy 1035 Burgundy 1108 Pink Carnation 1386 Eggplant 1354 Watermelon 1356 Pink Pearl 1081 Azalea 1141 Mauve 1186 Ruby Slipper 1382 Colonial Rose 1381 Ripe Raspberry 1236 Plum Brandy 3 4 CLASSIC RAYON 100% viscose While every attempt is made to reproduce thread colors accurately, colors on your monitor or print out may not precisely match thread colors. Color names are for your reference only. When ordering please refer to each color by color number. 1261 Lavendula 1198 Moonstone 1266 Regal Blue 1030 Light Periwinkle 1166 Hanukkah Blue 1364 Storm Sky Blue 1466 Sailor Blue 1365 Dusty Plum 1335 Dark Periwinkle