Genome-Wide Pooling Approach Identifies SPATA5 As a New Susceptibility Locus for Alopecia Areata Regina C
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -
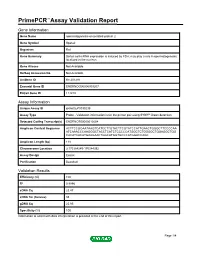
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name spermatogenesis-associated protein 2 Gene Symbol Spata2 Organism Rat Gene Summary Sertoli cell mRNA expression is induced by FSH; may play a role in spermatogenesis; localized to the nucleus Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Rn.201291 Ensembl Gene ID ENSRNOG00000009207 Entrez Gene ID 114210 Assay Information Unique Assay ID qRnoCEP0030238 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000012604 Amplicon Context Sequence ACTTCCGGAATAAGTCATCCTTGTACTTCGTATCCATTGAACTGGGCTTCCCCAA ATCAAACCCAAGGGCTACCTCATCTCCCCCATGGCTCTGGGGCTGGAGGCTGG CACATCACATGAAGAACTGGCATGGTGCCCATGGACCAGC Amplicon Length (bp) 118 Chromosome Location 3:170384245-170384392 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9996 cDNA Cq 23.47 cDNA Tm (Celsius) 85 gDNA Cq 25.95 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Spata2, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

4Q27 Deletion and 7Q36.1 Microduplication in a Patient With
Wu et al. BMC Medical Genomics (2020) 13:31 https://doi.org/10.1186/s12920-020-0697-y CASE REPORT Open Access 4q27 deletion and 7q36.1 microduplication in a patient with multiple malformations and hearing loss: a case report Maolan Wu, Xiangrong Zheng* , Xia Wang, Guoyuan Zhang and Jian Kuang Abstract Background: Chromosome deletions of the long arm of chromosome 4 in 4q syndrome are characterized by mild facial and digital dysmorphism, developmental delay, growth retardation, and skeletal and cardiac anomalies, which is regarded as an autism spectrum disorder. Moreover, some scarce reports indicate that patients with 4q interstitial deletion and 7p duplication may present symptoms associated with hearing loss. Case presentation: A boy with a severe developmental delay not only post-natal but also intrauterine and several dysmorphic features including microcephaly, ocular hypertelorism, exophthalmos, low-set ears, single palmar flexion crease, and overlapping toes presented discontinued cyanosis and recurrent respiratory infections. MRI, BAEP, echocardiogram and bronchoscopy revealed that he had persistent falcine sinus with a thin corpus callosum, left auditory pathway disorder, patent foramen ovale (2 mm), and tracheobronchomalacia with the right superior bronchus arising from the lateral posterior wall of the right main bronchus. Finally, the patient died with severe pneumonia at 10 months. Array CGH revealed a 23.62 Mb deletion at chromosome 4q27, arr [hg19] 4q27-q31.21 (121, 148, 089–144, 769, 263) × 1, and a 0.85 Mb duplication at chromosome 7q36.1, arr [hg19] 7q36.1-q36.2 (152, 510, 685–153, 363,5 98) × 3. It is rare for 4q syndrome cases or 7q duplications previously reported to have a hearing disorder, pulmonary dysplasia, and pulmonary arterial hypertension. -

Genetics of Amyotrophic Lateral Sclerosis in the Han Chinese
Genetics of amyotrophic lateral sclerosis in the Han Chinese Ji He A thesis submitted for the degree of Master of Philosophy at The University of Queensland in 2015 The University of Queensland Diamantina Institute 1 Abstract Amyotrophic lateral sclerosis is the most frequently occurring neuromuscular degenerative disorders, and has an obscure aetiology. Whilst major progress has been made, the majority of the genetic variation involved in ALS is, as yet, undefined. In this thesis, multiple genetic studies have been conducted to advance our understanding of the genetic architecture of the disease. In the light of the paucity of comprehensive genetic studies performed in Chinese, the presented study focused on advancing our current understanding in genetics of ALS in the Han Chinese population. To identify genetic variants altering risk of ALS, a genome-wide association study (GWAS) was performed. The study included 1,324 Chinese ALS cases and 3,115 controls. After quality control, a number of analyses were performed in a cleaned dataset of 1,243 cases and 2,854 controls that included: a genome-wide association analysis to identify SNPs associated with ALS; a genomic restricted maximum likelihood (GREML) analysis to estimate the proportion of the phenotypic variance in ALS liability due to common SNPs; and a gene- based analysis to identify genes associated with ALS. There were no genome-wide significant SNPs or genes associated with ALS. However, it was estimated that 17% (SE: 0.05; P=6×10-5) of the phenotypic variance in ALS liability was due to common SNPs. The top associated SNP was within GNAS (rs4812037; p =7×10-7). -

Mouse Spata2 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Spata2 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Spata2 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Spata2 gene (NCBI Reference Sequence: NM_170756 ; Ensembl: ENSMUSG00000047030 ) is located on Mouse chromosome 2. 3 exons are identified, with the ATG start codon in exon 2 and the TAG stop codon in exon 3 (Transcript: ENSMUST00000057627). Exon 2~3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Spata2 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP24-144E24 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Homozygous knockout leads to small testes, oligospermia, asthenozoospermia, reduced male fertility and decreased male germ cell numbers. It also affects necroptosis and increases inflammatory responses. Exon 2~3 covers 100.0% of the coding region. Start codon is in exon 2, and stop codon is in exon 3. The size of intron 1 for 5'-loxP site insertion: 7165 bp. The size of effective cKO region: ~2396 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy gRNA region Wildtype allele T A 5' gRNA region G 3' 1 2 3 Targeting vector T A G Targeted allele T A G Constitutive KO allele (After Cre recombination) Legends Exon of mouse Spata2 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Exomic Sequencing of Immune-Related Genes Reveals Novel Candidate Variants Associated with Alopecia Universalis
Exomic Sequencing of Immune-Related Genes Reveals Novel Candidate Variants Associated with Alopecia Universalis Seungbok Lee1,2., Seung Hwan Paik3., Hyun-Jin Kim1,2, Hyeong Ho Ryu3, Soeun Cha1, Seong Jin Jo3, Hee Chul Eun3,4,5, Jeong-Sun Seo1,2,6,7,8, Jong-Il Kim1,2,6,7*, Oh Sang Kwon3,4,5* 1 Genomic Medicine Institute (GMI), Medical Research Center, Seoul National University, Seoul, Korea, 2 Department of Biomedical Sciences, Seoul National University Graduate School, Seoul, Korea, 3 Department of Dermatology, Seoul National University College of Medicine, Seoul, Korea, 4 Laboratory of Cutaneous Aging and Hair Research, Clinical Research Institute, Seoul National University Hospital, Seoul, Korea, 5 Institute of Dermatological Science, Seoul National University College of Medicine, Seoul, Korea, 6 Department of Biochemistry and Molecular Biology, Seoul National University College of Medicine, Seoul, Korea, 7 Psoma Therapeutics Inc., Seoul, Korea, 8 Macrogen Inc., Seoul, Korea Abstract Alopecia areata (AA) is a common autoimmune disorder mostly presented as round patches of hair loss and subclassified into alopecia totalis/alopecia universalis (AT/AU) based on the area of alopecia. Although AA is relatively common, only 5% of AA patients progress to AT/AU, which affect the whole scalp and whole body respectively. To determine genetic determinants of this orphan disease, we undertook whole-exome sequencing of 6 samples from AU patients, and 26 variants in immune-related genes were selected as candidates. When an additional 14 AU samples were genotyped for these candidates, 6 of them remained at the level of significance in comparison with 155 Asian controls (p,1.9261023). -

Gene Ontology Functional Annotations and Pleiotropy
Network based analysis of genetic disease associations Sarah Gilman Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy under the Executive Committee of the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2013 Sarah Gilman All Rights Reserved ABSTRACT Network based analysis of genetic disease associations Sarah Gilman Despite extensive efforts and many promising early findings, genome-wide association studies have explained only a small fraction of the genetic factors contributing to common human diseases. There are many theories about where this “missing heritability” might lie, but increasingly the prevailing view is that common variants, the target of GWAS, are not solely responsible for susceptibility to common diseases and a substantial portion of human disease risk will be found among rare variants. Relatively new, such variants have not been subject to purifying selection, and therefore may be particularly pertinent for neuropsychiatric disorders and other diseases with greatly reduced fecundity. Recently, several researchers have made great progress towards uncovering the genetics behind autism and schizophrenia. By sequencing families, they have found hundreds of de novo variants occurring only in affected individuals, both large structural copy number variants and single nucleotide variants. Despite studying large cohorts there has been little recurrence among the genes implicated suggesting that many hundreds of genes may underlie these complex phenotypes. The question -

Rabbit Anti-SPATA5/FITC Conjugated Antibody
SunLong Biotech Co.,LTD Tel: 0086-571- 56623320 Fax:0086-571- 56623318 E-mail:[email protected] www.sunlongbiotech.com Rabbit Anti-SPATA5/FITC Conjugated antibody SL17631R-FITC Product Name: Anti-SPATA5/FITC Chinese Name: FITC标记的精子发生相关蛋白2抗体 AFG2; ATPase family gene 2 homolog; ATPase family protein 2 homolog; SPAF; Alias: Spermatogenesis associated 5; Spermatogenesis associated factor SPAF; Spermatogenesis-associated factor protein. Organism Species: Rabbit Clonality: Polyclonal React Species: Human,Mouse,Rat,Dog,Pig,Cow, ICC=1:50-200IF=1:50-200 Applications: not yet tested in other applications. optimal dilutions/concentrations should be determined by the end user. Molecular weight: 98kDa Form: Lyophilized or Liquid Concentration: 1mg/ml immunogen: KLH conjugated synthetic peptide derived from human SPATA5 Lsotype: IgG Purification: affinity purified by Protein A Storage Buffer: 0.01Mwww.sunlongbiotech.com TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol. Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year Storage: when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C. background: SPATA5 is an 893 amino acid protein that localizes to cytoplasm and mitochondrion, and may be involved in morphological and functional mitochondrial transformations during spermatogenesis. Existing as three alternatively spliced isoforms, SPATA5 Product Detail: belongs to the AAA ATPase family and the AFG2 subfamily. The gene that encodes SPATA5 consists of more than 396,000 bases and maps to human chromosome 4q28.1. -

Original Article Identification of Differentially Expressed Genes Between Male and Female Patients with Acute Myocardial Infarction Based on Microarray Data
Int J Clin Exp Med 2019;12(3):2456-2467 www.ijcem.com /ISSN:1940-5901/IJCEM0080626 Original Article Identification of differentially expressed genes between male and female patients with acute myocardial infarction based on microarray data Huaqiang Zhou1,2*, Kaibin Yang2*, Shaowei Gao1, Yuanzhe Zhang2, Xiaoyue Wei2, Zeting Qiu1, Si Li2, Qinchang Chen2, Yiyan Song2, Wulin Tan1#, Zhongxing Wang1# 1Department of Anesthesiology, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China; 2Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China. *Equal contributors and co-first au- thors. #Equal contributors. Received May 31, 2018; Accepted August 4, 2018; Epub March 15, 2019; Published March 30, 2019 Abstract: Background: Coronary artery disease has been the most common cause of death and the prognosis still needs further improving. Differences in the incidence and prognosis of male and female patients with coronary artery disease have been observed. We constructed this study hoping to understand those differences at the level of gene expression and to help establish gender-specific therapies. Methods: We downloaded the series matrix file of GSE34198 from the Gene Expression Omnibus database and identified differentially expressed genes between male and female patients. Gene ontology, Kyoto Encyclopedia of Genes and Genomes pathway enrichment analy- sis, and GSEA analysis of differentially expressed genes were performed. The protein-protein interaction network was constructed of the differentially expressed genes and the hub genes were identified. Results: A total of 215 up-regulated genes and 353 down-regulated genes were identified. The differentially expressed pathways were mainly related to the function of ribosomes, virus, and related immune response as well as the cell growth and proliferation. -

Loss-Of-Function Mutations in UDP-Glucose 6-Dehydrogenase Cause Recessive Developmental Epileptic Encephalopathy
ARTICLE https://doi.org/10.1038/s41467-020-14360-7 OPEN Loss-of-function mutations in UDP-Glucose 6-Dehydrogenase cause recessive developmental epileptic encephalopathy Holger Hengel et al.# Developmental epileptic encephalopathies are devastating disorders characterized by intractable epileptic seizures and developmental delay. Here, we report an allelic series of 1234567890():,; germline recessive mutations in UGDH in 36 cases from 25 families presenting with epileptic encephalopathy with developmental delay and hypotonia. UGDH encodes an oxidoreductase that converts UDP-glucose to UDP-glucuronic acid, a key component of specific proteogly- cans and glycolipids. Consistent with being loss-of-function alleles, we show using patients’ primary fibroblasts and biochemical assays, that these mutations either impair UGDH sta- bility, oligomerization, or enzymatic activity. In vitro, patient-derived cerebral organoids are smaller with a reduced number of proliferating neuronal progenitors while mutant ugdh zebrafish do not phenocopy the human disease. Our study defines UGDH as a key player for the production of extracellular matrix components that are essential for human brain development. Based on the incidence of variants observed, UGDH mutations are likely to be a frequent cause of recessive epileptic encephalopathy. #A full list of authors and their affiliations appears at the end of the paper. NATURE COMMUNICATIONS | (2020) 11:595 | https://doi.org/10.1038/s41467-020-14360-7 | www.nature.com/naturecommunications 1 ARTICLE NATURE COMMUNICATIONS | https://doi.org/10.1038/s41467-020-14360-7 evelopmental epileptic encephalopathies are a clinically A44V variant, identified in the Palestinian index family, was also Dand genetically heterogeneous group of devastating dis- found in two additional families from Puerto Rico (F11) and from orders characterized by severe epileptic seizures that are Spain (F13) indicative of independent but recurrent mutation in accompanied by developmental delay or regression1. -

The DNA Sequence and Comparative Analysis of Human Chromosome 20
articles The DNA sequence and comparative analysis of human chromosome 20 P. Deloukas, L. H. Matthews, J. Ashurst, J. Burton, J. G. R. Gilbert, M. Jones, G. Stavrides, J. P. Almeida, A. K. Babbage, C. L. Bagguley, J. Bailey, K. F. Barlow, K. N. Bates, L. M. Beard, D. M. Beare, O. P. Beasley, C. P. Bird, S. E. Blakey, A. M. Bridgeman, A. J. Brown, D. Buck, W. Burrill, A. P. Butler, C. Carder, N. P. Carter, J. C. Chapman, M. Clamp, G. Clark, L. N. Clark, S. Y. Clark, C. M. Clee, S. Clegg, V. E. Cobley, R. E. Collier, R. Connor, N. R. Corby, A. Coulson, G. J. Coville, R. Deadman, P. Dhami, M. Dunn, A. G. Ellington, J. A. Frankland, A. Fraser, L. French, P. Garner, D. V. Grafham, C. Grif®ths, M. N. D. Grif®ths, R. Gwilliam, R. E. Hall, S. Hammond, J. L. Harley, P. D. Heath, S. Ho, J. L. Holden, P. J. Howden, E. Huckle, A. R. Hunt, S. E. Hunt, K. Jekosch, C. M. Johnson, D. Johnson, M. P. Kay, A. M. Kimberley, A. King, A. Knights, G. K. Laird, S. Lawlor, M. H. Lehvaslaiho, M. Leversha, C. Lloyd, D. M. Lloyd, J. D. Lovell, V. L. Marsh, S. L. Martin, L. J. McConnachie, K. McLay, A. A. McMurray, S. Milne, D. Mistry, M. J. F. Moore, J. C. Mullikin, T. Nickerson, K. Oliver, A. Parker, R. Patel, T. A. V. Pearce, A. I. Peck, B. J. C. T. Phillimore, S. R. Prathalingam, R. W. Plumb, H. Ramsay, C. M.