Crystal Structure of the HLA-Cw3 Allotype-Specific Killer Cell Inhibitory Receptor KIR2DL2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Expression Tissue
The Journal of Immunology Killer Cell Ig-Like Receptor and Leukocyte Ig-Like Receptor Transgenic Mice Exhibit Tissue- and Cell-Specific Transgene Expression1 Danny Belkin,* Michaela Torkar,* Chiwen Chang,* Roland Barten,* Mauro Tolaini,† Anja Haude,* Rachel Allen,* Michael J. Wilson,* Dimitris Kioussis,† and John Trowsdale2* To generate an experimental model for exploring the function, expression pattern, and developmental regulation of human Ig-like activating and inhibitory receptors, we have generated transgenic mice using two human genomic clones: 52N12 (a 150-Kb clone encompassing the leukocyte Ig-like receptor (LILR)B1 (ILT2), LILRB4 (ILT3), and LILRA1 (LIR6) genes) and 1060P11 (a 160-Kb clone that contains ten killer cell Ig-like receptor (KIR) genes). Both the KIR and LILR families are encoded within the leukocyte receptor complex, and are involved in immune modulation. We have also produced a novel mAb to LILRA1 to facilitate expression studies. The LILR transgenes were expressed in a similar, but not identical, pattern to that observed in humans: LILRB1 was expressed in B cells, most NK cells, and a small number of T cells; LILRB4 was expressed in a B cell subset; and LILRA1 was found on a ring of cells surrounding B cell areas on spleen sections, consistent with other data showing monocyte/macrophage expression. KIR transgenic mice showed KIR2DL2 expression on a subset of NK cells and T cells, similar to the pattern seen in humans, and expression of KIR2DL4, KIR3DS1, and KIR2DL5 by splenic NK cells. These observations indicate that linked regulatory elements within the genomic clones are sufficient to allow appropriate expression of KIRs in mice, and illustrate that the presence of the natural ligands for these receptors, in the form of human MHC class I proteins, is not necessary for the expression of the KIRs observed in these mice. -

Phyre 2 Results for A1YIY0
Email [email protected] Description A1YIY0 Tue Jul 30 13:19:15 BST Date 2013 Unique Job 1035bc4b501530df ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:cell adhesion Chain: A: PDB Molecule:down syndrome cell adhesion molecule 1 c3dmkA_ Alignment 100.0 14 (dscam) isoform PDBTitle: crystal structure of down syndrome cell adhesion molecule (dscam)2 isoform 1.30.30, n-terminal eight ig domains PDB header:cell adhesion 2 c2om5A_ Alignment 100.0 20 Chain: A: PDB Molecule:contactin 2; PDBTitle: n-terminal fragment of human tax1 PDB header:cell adhesion 3 c3jxaA_ Alignment 100.0 21 Chain: A: PDB Molecule:contactin 4; PDBTitle: immunoglobulin domains 1-4 of mouse cntn4 PDB header:signaling protein/transferase Chain: A: PDB Molecule:tek tyrosine kinase variant; 4 c4k0vA_ 100.0 12 Alignment PDBTitle: structural basis for angiopoietin-1 mediated signaling initiation PDB header:immune system Chain: A: PDB Molecule:natural cytotoxicity triggering receptor 1; 5 c1p6fA_ 100.0 9 Alignment PDBTitle: structure of the human natural cytotoxicity receptor nkp46 PDB header:immune system/receptor 6 c1ollA_ Alignment 99.9 9 Chain: A: PDB Molecule:nk receptor; PDBTitle: extracellular region of the human receptor nkp46 PDB header:viral protein Chain: A: PDB Molecule:hoc head outer capsid protein; 7 c3shsA_ 99.9 18 Alignment PDBTitle: three n-terminal domains of the bacteriophage rb49 highly immunogenic2 outer capsid protein (hoc) PDB header:immune system Chain: E: PDB Molecule:natural -

Differential Integrin Adhesome Expression Defines Human Natural
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.01.404806; this version posted December 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Differential integrin adhesome expression defines human natural killer cell residency and developmental stage Everardo Hegewisch Solloa1, Seungmae Seo1, Bethany L. Mundy-Bosse2, Anjali Mishra3,a, Erik Waldman4,b, Sarah Maurrasse4,b, Eli Grunstein4, Thomas J. Connors5, Aharon G. Freud2,6, and Emily M. Mace1 1Department of Pediatrics, Columbia University College of Physicians and Surgeons, New York NY 10032 2Division of Hematology, Department of Internal Medicine, The Ohio State University, Columbus, OH 43210, USA; Comprehensive Cancer Center and The James Cancer Hospital and Solove Research Institute, The Ohio State University, Columbus, OH 43210 33Division of Dermatology, Department of Internal Medicine, The Ohio State University, Columbus, OH 43210, USA; Comprehensive Cancer Center and The James Cancer Hospital and Solove Research Institute, The Ohio State University, Columbus, OH 43210 4Department of Otolaryngology - Head and Neck Surgery, Columbia University Medical Center, New York, New York 10032 5Department of Pediatrics, Division of Pediatric Critical Care and Hospital Medicine, Columbia University Irving Medical Center, New York, NY 10024 6Department of Pathology, The Ohio State University, Columbus, -

Snipa Snpcard
SNiPAcard Block annotations Block info genomic range chr19:55,117,749-55,168,602 block size 50,854 bp variant count 74 variants Basic features Conservation/deleteriousness Linked genes μ = -0.557 [-4.065 – AC009892.10 , AC009892.5 , AC009892.9 , AC245036.1 , AC245036.2 , phyloP 2.368] gene(s) hit or close-by AC245036.3 , AC245036.4 , AC245036.5 , AC245036.6 , LILRA1 , LILRB1 , LILRB4 , MIR8061 , VN1R105P phastCons μ = 0.059 [0 – 0.633] eQTL gene(s) CTB-83J4.2 , LILRA1 , LILRA2 , LILRB2 , LILRB5 , LILRP1 μ = -0.651 [-4.69 – 2.07] AC008984.5 , AC008984.5 , AC008984.6 , AC008984.6 , AC008984.7 , AC008984.7 , AC009892.10 , AC009892.10 , AC009892.2 , AC009892.2 , AC009892.5 , AC010518.3 , AC010518.3 , AC011515.2 , AC011515.2 , AC012314.19 , AC012314.19 , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , FCAR , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL1 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL3 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , KIR2DL4 , -

Cytolytic Activity of NK Cell Clones Against Acute Childhood Precursor
Bone Marrow Transplantation (2009) 43, 875–881 & 2009 Macmillan Publishers Limited All rights reserved 0268-3369/09 $32.00 www.nature.com/bmt ORIGINAL ARTICLE Cytolytic activity of NK cell clones against acute childhood precursor-B-cell leukaemia is influenced by HLA class I expression on blasts and the differential KIR phenotype of NK clones T Feuchtinger1, M Pfeiffer1, A Pfaffle1, H-M Teltschik1, D Wernet2, M Schumm1, R Lotfi2, R Handgretinger1 and P Lang1 1Department of Paediatric Haematology/Oncology, University Children’s Hospital, Tu¨bingen, Germany and 2Institute of Transfusion Medicine, Eberhard Karls University, Tu¨bingen, Germany Relapse after allo-SCT in patients with acute leukaemia Introduction remains a major problem. A beneficial impact of alloreactive natural killer (NK) cells has been reported Stem cell transplantation from matched or partially for myeloid malignancies, but has been questionable for matched unrelated donors as well as mismatched related B-lineage ALL. We analysed lysis of primary paediatric donors has become an established procedure for the precursor-B-ALL blasts by 285 NK cell clones to treatment of children with high risk and relapsed leukae- investigate whether HLA class I expression on the blasts mia.1–3 GVHD-related mortality has been reduced signifi- and phenotypic killer cell Ig-like receptor (KIR) expres- cantly by T-cell depletion, but relapse still remains the sion on NK cells affect the lytic activity against ALL major problem after successful transplantation.4,5 In blasts. Precursor-B-ALL blasts with low HLA-I expres- contrast to myeloid malignancies, controversial effects of sion were lysed by a majority (79%) of NK cell clones, T-cell depletion on relapse rate in ALL have been whereas those with high HLA-I expression showed low reported.1,3,6,7 As normal numbers of NK cells reconstitute susceptibility to NK clones independent of their KIR from donor progenitors within a few weeks after T-cell expression patterns. -

High-Dimensional Analysis Reveals a Distinct Population of Adaptive-Like Tissue
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 High-dimensional analysis reveals a distinct population of adaptive-like tissue- 2 resident NK cells in human lung 3 4 Nicole Marquardt1*, Marlena Scharenberg1, Jeffrey E. Mold2, Joanna Hård2, Eliisa 5 Kekäläinen3,4, Marcus Buggert1, Son Nguyen5,6, Jennifer N. Wilson1, Mamdoh Al- 6 Ameri7, Hans-Gustaf Ljunggren1, Jakob Michaëlsson1 7 8 Running title: Adaptive-like human lung tissue-resident NK cells 9 10 Affiliations: 11 1Center for Infectious Medicine, Department of Medicine, Karolinska Institutet, 12 Stockholm, Sweden 13 2Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden 14 3Translational Immunology Research Program & Department of Bacteriology and 15 Immunology, University of Helsinki, Finland 16 4HUSLAB, Division of Clinical Microbiology, Helsinki University Hospital, Helsinki, 17 Finland, 18 5Department of Microbiology, Perelman School of Medicine, University of 19 Pennsylvania, Philadelphia, PA, USA 20 6Institute for Immunology, Perelman School of Medicine, University of Pennsylvania, 21 Philadelphia, PA, USA 22 7Thoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska 23 Institutet, Karolinska University Hospital, Stockholm, Sweden 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
Type of the Paper (Article
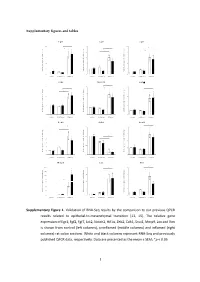
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

Natural Killer Cell Lymphoma Shares Strikingly Similar Molecular Features
Leukemia (2011) 25, 348–358 & 2011 Macmillan Publishers Limited All rights reserved 0887-6924/11 www.nature.com/leu ORIGINAL ARTICLE Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic cd T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro J Iqbal1, DD Weisenburger1, A Chowdhury2, MY Tsai2, G Srivastava3, TC Greiner1, C Kucuk1, K Deffenbacher1, J Vose4, L Smith5, WY Au3, S Nakamura6, M Seto6, J Delabie7, F Berger8, F Loong3, Y-H Ko9, I Sng10, X Liu11, TP Loughran11, J Armitage4 and WC Chan1, for the International Peripheral T-cell Lymphoma Project 1Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE, USA; 2Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE, USA; 3Departments of Pathology and Medicine, University of Hong Kong, Queen Mary Hospital, Hong Kong, China; 4Division of Hematology and Oncology, Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA; 5College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA; 6Departments of Pathology and Cancer Genetics, Aichi Cancer Center Research Institute, Nagoya University, Nagoya, Japan; 7Department of Pathology, University of Oslo, Norwegian Radium Hospital, Oslo, Norway; 8Department of Pathology, Centre Hospitalier Lyon-Sud, Lyon, France; 9Department of Pathology, Samsung Medical Center, Sungkyunkwan University, Seoul, Korea; 10Department of Pathology, Singapore General Hospital, Singapore and 11Penn State Hershey Cancer Institute, Pennsylvania State University College of Medicine, Hershey, PA, USA Natural killer (NK) cell lymphomas/leukemias are rare neo- Introduction plasms with an aggressive clinical behavior. -

Restoring Natural Killer Cell Immunity Against Multiple Myeloma in the Era of New Drugs
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Institutional Research Information System University of Turin Restoring Natural Killer Cell immunity against Multiple Myeloma in the era of New Drugs Gianfranco Pittari1, Luca Vago2,3, Moreno Festuccia4,5, Chiara Bonini6,7, Deena Mudawi1, Luisa Giaccone4,5 and Benedetto Bruno4,5* 1 Department of Medical Oncology, National Center for Cancer Care and Research, HMC, Doha, Qatar, 2 Unit of Immunogenetics, Leukemia Genomics and Immunobiology, IRCCS San Raffaele Scientific Institute, Milano, Italy, 3 Hematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milano, Italy, 4 Department of Oncology/Hematology, A.O.U. Città della Salute e della Scienza di Torino, Presidio Molinette, Torino, Italy, 5 Department of Molecular Biotechnology and Health Sciences, University of Torino, Torino, Italy, 6 Experimental Hematology Unit, Division of Immunology, Transplantation and Infectious Diseases, IRCCS San Raffaele Scientific Institute, Milano, Italy, 7 Vita-Salute San Raffaele University, Milano, Italy Transformed plasma cells in multiple myeloma (MM) are susceptible to natural killer (NK) cell-mediated killing via engagement of tumor ligands for NK activating receptors or “missing-self” recognition. Similar to other cancers, MM targets may elude NK cell immunosurveillance by reprogramming tumor microenvironment and editing cell surface antigen repertoire. Along disease continuum, these effects collectively result in a pro- gressive decline of NK cell immunity, a phenomenon increasingly recognized as a critical determinant of MM progression. In recent years, unprecedented efforts in drug devel- opment and experimental research have brought about emergence of novel therapeutic interventions with the potential to override MM-induced NK cell immunosuppression. -

KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV
KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV This information is current as Jeanette E. Boudreau, Tiernan J. Mulrooney, Jean-Benoît Le of September 25, 2021. Luduec, Edward Barker and Katharine C. Hsu J Immunol 2016; 196:3398-3410; Prepublished online 9 March 2016; doi: 10.4049/jimmunol.1502469 http://www.jimmunol.org/content/196/8/3398 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2016/03/08/jimmunol.150246 Material 9.DCSupplemental http://www.jimmunol.org/ References This article cites 72 articles, 37 of which you can access for free at: http://www.jimmunol.org/content/196/8/3398.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 25, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology KIR3DL1 and HLA-B Density and Binding Calibrate NK Education and Response to HIV Jeanette E. -

KIR3DL1 and HLA-A and HLA-B Peptide-Dependent Interactions Between Cutting Edge: Allele-Specific
Cutting Edge: Allele-Specific and Peptide-Dependent Interactions between KIR3DL1 and HLA-A and HLA-B This information is current as Hathairat Thananchai, Geraldine Gillespie, Maureen P. of September 29, 2021. Martin, Arman Bashirova, Nobuyo Yawata, Makoto Yawata, Philippa Easterbrook, Daniel W. McVicar, Katsumi Maenaka, Peter Parham, Mary Carrington, Tao Dong and Sarah Rowland-Jones J Immunol 2007; 178:33-37; ; Downloaded from doi: 10.4049/jimmunol.178.1.33 http://www.jimmunol.org/content/178/1/33 References This article cites 28 articles, 12 of which you can access for free at: http://www.jimmunol.org/ http://www.jimmunol.org/content/178/1/33.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 29, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2007 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. THE JOURNAL OF IMMUNOLOGY CUTTING EDGE Cutting Edge: Allele-Specific and Peptide-Dependent Interactions between KIR3DL1 and HLA-A and HLA-B1,2 † ‡ Hathairat Thananchai,* Geraldine Gillespie,* Maureen P. -

Recombinant Proteins & Monoclonal Antibodies
www.nkmaxbio.com NK CELL Recombinant Proteins & Monoclonal Antibodies "에이티젠"이 "엔케이맥스"로 새롭게 시작합니다. Products for NK Cell Surface Receptor Natural killer cells or NK cells are a type of cytotoxic lymphocyte critical to the innate immune system. NK cell receptors can also be differentiated based on function. Natural killer cell activation is determined by the balance of inhibitory and activating receptor stimulation. Key receptors on human NK cells KIR2DS4 HLA-C NKG2C HLA-E Activating NKG2D MIC-A/B, ULBP Receptor CD16 lgG NKp30 B7-H6 NKp44 Viral HA KIR2DL1 HLA-C NKG2A HLA-E NK cell Inhibitory Target cell KIR3DL1 HLA-BW4, HLA-A Receptor TIGIT CD112, CD113, CD155 KLRG1 Cadherins DNAM-1 CD112, CD155 NTB-A NTB-A/SLAMF6 Coreceptor LAIR-1 Collagen 2B4 CD48 Related Monoclonal Antibodies Product name Cat No. Clone No. Applications Isotype Host NKp44 ATGA0322 AT1G6 ELISA, WB, FACS, ICC/IF IgG2a, k M KIR2DS4 AKR0504 5F2 ELISA, WB IgG2b, k M KLRD1 ATGA0487 AT13E3 ELISA, WB, FACS IgG1, k M Activating NKp46 AKR0408 n1D9 ELISA, WB, FACS IgG1, k M KIR2DL1 AKR0620 2F9 ELISA, WB, FACS, ICC/IF IgG2a, k M KIR2DL3 AKR3006 190IIC311 ELISA, WB, ICC/IF IgG2a, k M Inhibitory KIR2DL4 AKR0501 2H6 ELISA, WB IgG2b, k M KIR2DL1 antibody(2F9) KIR2DL3 antibody(190IIC311) NKp46 antibody(n1D9) Cat No. AKR0620 Cat No. AKR3006 Cat No. AKR0408 ICC/IF analysis ICC/IF analysis Flow Cytometry ICC/IF analysis of KIR2DL1 in Hep3B ICC/IF analysis of KIR2DL3 in HeLa Flow cytometry analysis of NKp46 in cells line, stained with DAPI (Blue) for cells line, stained with DAPI (Blue) for HeLa cell, staining at 2-5μg for 1x106 nucleus staining and monoclonal nucleus staining and monoclonal cells (Red) with mouse anti-lgG (Blue), anti-human KIR2DL1 antibody (1:100) anti-human KIR2DL3 antibody (1:100) goat anti-mouse lgG-Alexa flour 488.