Fibroblast Growth Factor 2-Mediated Cardioprotection: the Kinase Mediators and Downstream Targets of FGF2-Induced Protection from Ischemia and Reperfusion Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pnas11138correction 14002..14003
Corrections MEDICAL SCIENCES Correction for “Regulation of bone remodeling by vasopressin New, Alberta Zallone, and Mone Zaidi, which appeared in issue 46, explains the bone loss in hyponatremia,” by Roberto Tamma, November 12, 2013, of Proc Natl Acad Sci USA (110:18644–18649; Li Sun, Concetta Cuscito, Ping Lu, Michelangelo Corcelli, first published October 28, 2013; 10.1073/pnas.1318257110). Jianhua Li, Graziana Colaianni, Surinder S. Moonga, Adriana The authors note that Fig. 1 appeared incorrectly. The cor- Di Benedetto, Maria Grano, Silvia Colucci, Tony Yuen, Maria I. rected figure and its legend appear below. Fig. 1. Bone cells express Avprs. Immunofluorescence micrographs (A) and Western immunoblotting (B) show the expression of Avpr1α in osteoblasts and osteoclasts, and as a function of osteoblast (mineralization) and osteoclast (with Rankl) differentiation. The expression of Avp (ligand) and Avpr1α (receptor) in osteoblasts is regulated by 17β-estradiol, as determined by quantitative PCR (C) and Western immunoblotting (D). (Magnification: A,63×.) Because Avp is a small peptide, its precursor neurophysin II is measured. Statistics: Student t test, P values shown compared with 0 h. Stimulation of Erk phosphorylation − (p-Erk) as a function of total Erk (t-Erk) by Avp (10 8 M) in osteoclast precursors (preosteoclasts), osteoclasts (OC), and osteoblasts establishes functionality of − the Avpr1α in the presence or absence of the receptor inhibitor SR49059 (10 8 M) (E). Western immunoblotting showing the expression of Avpr2 in pre- −/− osteoclasts, OCs (F), and osteoblasts (G) isolated from Avpr1α mice, as well as in MC3T3.E1 osteoblast precursors (G). Functionality of Avpr2 was confirmed −/− by the demonstration that cells from Avpr1α mice remained responsive to AVP in reducing the expression of osteoblast differentiation genes, namely Runx2, Osx, Bsp, Atf4, Opn, and Osteocalcin (quantitative PCR, P values shown) (H). -

Sat-196 How to Estimate Glomerular Filtration Rate
ISN WCN 2019 ABSTRACTS 13 (28%) patients had CKD stage 3. 6/13 had low predicted risk of Nephrology, Melbourne, Australia, 8Austin Hospital, Nephrology, Heidel- berg, Australia, 9Monash Childrens Hospital, Nephrology, Australia, progression at 5 years of whom 4/6 progressed unexpectedly. 1/13 was 10 identified as having high risk at 5 years and that 1 patient progressed to Melbourne Genomics Health Alliance, Victorian Clinical Genetics Service, Melbourne, Australia, 11Australian Genomic Health Alliance, KidGen Renal CKD5D/T. Genetics Flagship, Australia, Australia, 12Royal Childrens Hospital, 6/18 CKD 3/4 patients with predicted low risk progressed to CKD5D/ Nephrology, Melbourne, Australia T unexpectedly. 1/6 had emergency abdominal surgery, 1/6 patient had Introduction: Genomic technologies enable the rapid and cost-effective unexplained rapid progression and 4/6 had acute upper gastro-intes- fi tinal haemorrhage causing terminal decline of kidney function. sequencing of DNA and have demonstrated a de nitive diagnosis in Conclusions: The number of patients analysed was small. The 8-vari- several patient groups. The clinical utility of whole exome sequencing able equation accurately predicted high risk of progression to CKD5D/T (WES) in a kidney disease cohort is not yet well established. We in 7/9 CKD3/4 patients. describe the patient characteristics and diagnostic yield of a cohort of Conversely, 6/18 patients with predicted low risk progressed to 200 patients with suspected genetic kidney disease referred for WES via CKD5D/T. Acute medical events including upper gastro-intestinal bleed a multidisciplinary renal genetics clinic. Methods: accounted for most instances of unexpected progression. 200 sequential patients were recruited into a prospective observational cohort study through five tertiary academic centres in Victoria, Australia. -

The Role of Z-Disc Proteins in Myopathy and Cardiomyopathy
International Journal of Molecular Sciences Review The Role of Z-disc Proteins in Myopathy and Cardiomyopathy Kirsty Wadmore 1,†, Amar J. Azad 1,† and Katja Gehmlich 1,2,* 1 Institute of Cardiovascular Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham B15 2TT, UK; [email protected] (K.W.); [email protected] (A.J.A.) 2 Division of Cardiovascular Medicine, Radcliffe Department of Medicine and British Heart Foundation Centre of Research Excellence Oxford, University of Oxford, Oxford OX3 9DU, UK * Correspondence: [email protected]; Tel.: +44-121-414-8259 † These authors contributed equally. Abstract: The Z-disc acts as a protein-rich structure to tether thin filament in the contractile units, the sarcomeres, of striated muscle cells. Proteins found in the Z-disc are integral for maintaining the architecture of the sarcomere. They also enable it to function as a (bio-mechanical) signalling hub. Numerous proteins interact in the Z-disc to facilitate force transduction and intracellular signalling in both cardiac and skeletal muscle. This review will focus on six key Z-disc proteins: α-actinin 2, filamin C, myopalladin, myotilin, telethonin and Z-disc alternatively spliced PDZ-motif (ZASP), which have all been linked to myopathies and cardiomyopathies. We will summarise pathogenic variants identified in the six genes coding for these proteins and look at their involvement in myopathy and cardiomyopathy. Listing the Minor Allele Frequency (MAF) of these variants in the Genome Aggregation Database (GnomAD) version 3.1 will help to critically re-evaluate pathogenicity based on variant frequency in normal population cohorts. -

Ever 2018 Eupo 2018
European Association for Vision and Eye Research European University Professors of Ophthalmology EVER 2018 Annual Congress October 4-6, 2018 EUPO 2018 Course on Retina, Intraocular Inflammation & Uveitis October 3-4, 2018 Programme book Nice, France www.ever.be www.eupo.eu European Association for Vision and Eye Research EVER 20October 17-1919 in Nice, France www.ever.be 1 Table of contents Word from the president ....................................................................................................................................2 About EVER ..........................................................................................................................................................3 EVER Membership ...............................................................................................................................................4 Speakers’ affiliation to scientific sections .........................................................................................................5 Composition of the board 2018 .........................................................................................................................8 Venue ................................................................................................................................................................... 10 Congress information ....................................................................................................................................... 11 Programme information ....................................................................................................................................15 -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

MAP2K3 (Human) Recombinant Protein (Q01)
MAP2K3 (Human) Recombinant phosphorylates and thus activates MAPK14/p38-MAPK. Protein (Q01) This kinase can be activated by insulin, and is necessary for the expression of glucose transporter. Expression of Catalog Number: H00005606-Q01 RAS oncogene is found to result in the accumulation of the active form of this kinase, which thus leads to the Regulation Status: For research use only (RUO) constitutive activation of MAPK14, and confers oncogenic transformation of primary cells. The inhibition Product Description: Human MAP2K3 partial ORF ( of this kinase is involved in the pathogenesis of Yersina AAH32478, 1 a.a. - 100 a.a.) recombinant protein with pseudotuberculosis. Multiple alternatively spliced GST-tag at N-terminal. transcript variants that encode distinct isoforms have been reported for this gene. [provided by RefSeq] Sequence: MESPASSQPASMPQSKGKSKRKKDLRISCMSKPPAP NPTPPRNLDSRTFITIGDRNFEVEADDLVTISELGRGAY GVVEKVRHAQSGTIMAVKRIRATVN Host: Wheat Germ (in vitro) Theoretical MW (kDa): 36.63 Applications: AP, Array, ELISA, WB-Re (See our web site product page for detailed applications information) Protocols: See our web site at http://www.abnova.com/support/protocols.asp or product page for detailed protocols Preparation Method: in vitro wheat germ expression system Purification: Glutathione Sepharose 4 Fast Flow Storage Buffer: 50 mM Tris-HCI, 10 mM reduced Glutathione, pH=8.0 in the elution buffer. Storage Instruction: Store at -80°C. Aliquot to avoid repeated freezing and thawing. Entrez GeneID: 5606 Gene Symbol: MAP2K3 Gene Alias: MAPKK3, MEK3, MKK3, PRKMK3 Gene Summary: The protein encoded by this gene is a dual specificity protein kinase that belongs to the MAP kinase kinase family. This kinase is activated by mitogenic and environmental stress, and participates in the MAP kinase-mediated signaling cascade. -

RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases
G C A T T A C G G C A T genes Review RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases Amber Willbanks, Shaun Wood and Jason X. Cheng * Department of Pathology, Hematopathology Section, University of Chicago, Chicago, IL 60637, USA; [email protected] (A.W.); [email protected] (S.W.) * Correspondence: [email protected] Abstract: Chromatin structure plays an essential role in eukaryotic gene expression and cell identity. Traditionally, DNA and histone modifications have been the focus of chromatin regulation; however, recent molecular and imaging studies have revealed an intimate connection between RNA epigenetics and chromatin structure. Accumulating evidence suggests that RNA serves as the interplay between chromatin and the transcription and splicing machineries within the cell. Additionally, epigenetic modifications of nascent RNAs fine-tune these interactions to regulate gene expression at the co- and post-transcriptional levels in normal cell development and human diseases. This review will provide an overview of recent advances in the emerging field of RNA epigenetics, specifically the role of RNA modifications and RNA modifying proteins in chromatin remodeling, transcription activation and RNA processing, as well as translational implications in human diseases. Keywords: 5’ cap (5’ cap); 7-methylguanosine (m7G); R-loops; N6-methyladenosine (m6A); RNA editing; A-to-I; C-to-U; 2’-O-methylation (Nm); 5-methylcytosine (m5C); NOL1/NOP2/sun domain Citation: Willbanks, A.; Wood, S.; (NSUN); MYC Cheng, J.X. RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases. Genes 2021, 12, 627. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Application of a MYC Degradation
SCIENCE SIGNALING | RESEARCH ARTICLE CANCER Copyright © 2019 The Authors, some rights reserved; Application of a MYC degradation screen identifies exclusive licensee American Association sensitivity to CDK9 inhibitors in KRAS-mutant for the Advancement of Science. No claim pancreatic cancer to original U.S. Devon R. Blake1, Angelina V. Vaseva2, Richard G. Hodge2, McKenzie P. Kline3, Thomas S. K. Gilbert1,4, Government Works Vikas Tyagi5, Daowei Huang5, Gabrielle C. Whiten5, Jacob E. Larson5, Xiaodong Wang2,5, Kenneth H. Pearce5, Laura E. Herring1,4, Lee M. Graves1,2,4, Stephen V. Frye2,5, Michael J. Emanuele1,2, Adrienne D. Cox1,2,6, Channing J. Der1,2* Stabilization of the MYC oncoprotein by KRAS signaling critically promotes the growth of pancreatic ductal adeno- carcinoma (PDAC). Thus, understanding how MYC protein stability is regulated may lead to effective therapies. Here, we used a previously developed, flow cytometry–based assay that screened a library of >800 protein kinase inhibitors and identified compounds that promoted either the stability or degradation of MYC in a KRAS-mutant PDAC cell line. We validated compounds that stabilized or destabilized MYC and then focused on one compound, Downloaded from UNC10112785, that induced the substantial loss of MYC protein in both two-dimensional (2D) and 3D cell cultures. We determined that this compound is a potent CDK9 inhibitor with a previously uncharacterized scaffold, caused MYC loss through both transcriptional and posttranslational mechanisms, and suppresses PDAC anchorage- dependent and anchorage-independent growth. We discovered that CDK9 enhanced MYC protein stability 62 through a previously unknown, KRAS-independent mechanism involving direct phosphorylation of MYC at Ser . -

Short-Term Rapamycin Persistently Improves Cardiac Function After Cessation of Treatment in Aged Male and Female Mice
Short-term rapamycin persistently improves cardiac function after cessation of treatment in aged male and female mice. Ellen Quarles A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2017 Reading Committee: Peter Rabinovitch, Chair Michael MacCoss David Marcinek Program Authorized to Offer Degree: Pathology © Copyright 2017 Ellen Quarles University of Washington Abstract Short-term rapamycin persistently improves cardiac function after cessation of treatment in aged male and female mice. Ellen Quarles Chair of the Supervisory Committee: Peter Rabinovitch, Professor and Vice Chair of Research Department of Pathology Cardiac aging is an intrinsic process that results in impaired cardiac function and dysregulation of cellular and molecular quality control mechanisms. These effects are evident in the decline of diastolic function, increase in left ventricular hypertrophy, metabolic substrate shifts, and alterations to the cardiac proteome. This thesis covers the quality control mechanisms that are associated with cardiac aging, results from an anti-aging intervention in aged mice, and a review of mitochondrial dysfunction in the heart. Chapter one is a review of the quality control mechanisms in aging myocardium. Chapter two consists of the results of several mouse experiments that compare the cardiac function, proteomes, and metabolomes of aged and young controls, along with rapamycin treated aged mice. The novelty of this study comes from the inclusion of a group of animals treated only transiently with the drug, then followed for eight weeks post-drug-removal. This persistence cohort may hold clues to deriving long-lasting benefits of rapamycin with only transient treatment. -
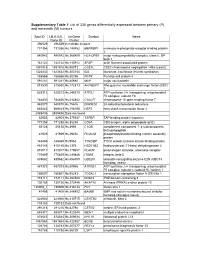
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator,