Using Dynamic Combinatorial Chemistry to Construct Novel Thioester and Disulfide Lipids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

De Novo Biosynthesis of Terminal Alkyne-Labeled Natural Products
De novo biosynthesis of terminal alkyne-labeled natural products Xuejun Zhu1,2, Joyce Liu2,3, Wenjun Zhang1,2,4* 1Department of Chemical and Biomolecular Engineering, 2Energy Biosciences Institute, 3Department of Bioengineering, University of California, Berkeley, CA 94720, USA. 4Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. *e-mail:[email protected] 1 Abstract: The terminal alkyne is a functionality widely used in organic synthesis, pharmaceutical science, material science, and bioorthogonal chemistry. This functionality is also found in acetylenic natural products, but the underlying biosynthetic pathways for its formation are not well understood. Here we report the characterization of the first carrier protein- dependent terminal alkyne biosynthetic machinery in microbes. We further demonstrate that this enzymatic machinery can be exploited for the in situ generation and incorporation of terminal alkynes into two natural product scaffolds in E. coli. These results highlight the prospect for tagging major classes of natural products, including polyketides and polyketide/non-ribosomal peptide hybrids, using biosynthetic pathway engineering. 2 Natural products are important small molecules widely used as drugs, pesticides, herbicides, and biological probes. Tagging natural products with a unique chemical handle enables the visualization, enrichment, quantification, and mode of action study of natural products through bioorthogonal chemistry1-4. One prevalent bioorthogonal reaction is -

Phospholipid:Diacylglycerol Acyltransferase: an Enzyme That Catalyzes the Acyl-Coa-Independent Formation of Triacylglycerol in Yeast and Plants
Phospholipid:diacylglycerol acyltransferase: An enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants Anders Dahlqvist*†‡, Ulf Ståhl†§, Marit Lenman*, Antoni Banas*, Michael Lee*, Line Sandager¶, Hans Ronne§, and Sten Stymne¶ *Scandinavian Biotechnology Research (ScanBi) AB, Herman Ehles Va¨g 2 S-26831 Svaloˆv, Sweden; ¶Department of Plant Breeding Research, Swedish University of Agricultural Sciences, Herman Ehles va¨g 2–4, S-268 31 Svalo¨v, Sweden; and §Department of Plant Biology, Uppsala Genetic Center, Swedish University of Agricultural Sciences, Box 7080, S-750 07 Uppsala, Sweden Edited by Christopher R. Somerville, Carnegie Institution of Washington, Stanford, CA, and approved March 31, 2000 (received for review February 15, 2000) Triacylglycerol (TAG) is known to be synthesized in a reaction that acid) and epoxidated fatty acid (vernolic acid) in TAG in castor uses acyl-CoA as acyl donor and diacylglycerol (DAG) as acceptor, bean (Ricinus communis) and the hawk’s-beard Crepis palaestina, and which is catalyzed by the enzyme acyl-CoA:diacylglycerol respectively. Furthermore, a similar enzyme is shown to be acyltransferase. We have found that some plants and yeast also present in the yeast Saccharomyces cerevisiae, and the gene have an acyl-CoA-independent mechanism for TAG synthesis, encoding this enzyme, YNR008w, is identified. which uses phospholipids as acyl donors and DAG as acceptor. This reaction is catalyzed by an enzyme that we call phospholipid:dia- Materials and Methods cylglycerol acyltransferase, or PDAT. PDAT was characterized in Yeast Strains and Plasmids. The wild-type yeast strains used were microsomal preparations from three different oil seeds: sunflower, either FY1679 (MAT␣ his3-⌬200 leu2-⌬1 trp1-⌬6 ura3-52) (9) or castor bean, and Crepis palaestina. -

Alkyldihydropyrones, New Polyketides Synthesized by a Type III Polyketide Synthase from Streptomyces Reveromyceticus
The Journal of Antibiotics (2014) 67, 819–823 & 2014 Japan Antibiotics Research Association All rights reserved 0021-8820/14 www.nature.com/ja ORIGINAL ARTICLE Alkyldihydropyrones, new polyketides synthesized by a type III polyketide synthase from Streptomyces reveromyceticus Teruki Aizawa1, Seung-Young Kim1, Shunji Takahashi2,3, Masahiko Koshita1, Mioka Tani1, Yushi Futamura3, Hiroyuki Osada2,3 and Nobutaka Funa1 Genome sequencing allows a rapid and efficient identification of novel catalysts that produce novel secondary metabolites. Here we describe the catalytic properties of dihydropyrone synthase A (DpyA), a novel type III polyketide synthase encoded in a linear plasmid of Streptomyces reveromyceticus. Heterologous expression of dpyA led to the accumulation of alkyldihydropyrones A (1), B (2), C (3) and D (4), which are novel dihydropyran compounds that exhibit weak cytotoxicity against the leukemia cell line HL-60. DpyA catalyzes the condensation of b-hydroxyl acid thioester and methylmalonyl-CoA to yield a triketide intermediate that then undergoes lactonization of a secondary alcohol and a thioester to give alkyldihydropyrone. The Journal of Antibiotics (2014) 67, 819–823; doi:10.1038/ja.2014.80; published online 2 July 2014 INTRODUCTION Very recently, Tang et al.11 discovered that presulficidin, which is Type III polyketide synthase (PKS), which is widely distributed synthesized by Cpz6, a type III PKS from the caprazamycin among higher plants, fungi and bacteria, catalyzes the assembly of biosynthetic cluster, relays sulfonate from 30-phosphoadenine-50- primary metabolites such as acyl-CoA compounds to synthesize phosphosulfate to caprazamycin. structurally complex polyketides.1 The reaction catalyzed by type III Genome sequence analyses of Streptomyces species have shown that PKS starts with the formation of a thioester bond between the the number of biosynthetic gene clusters encoded on the chromosome catalytic center cysteine and an acyl group derived from a starter is much higher than the number of secondary metabolites isolated substrate. -

The Reaction of Aminonitriles with Aminothiols: a Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean
life Article The Reaction of Aminonitriles with Aminothiols: A Way to Thiol-Containing Peptides and Nitrogen Heterocycles in the Primitive Earth Ocean Ibrahim Shalayel , Seydou Coulibaly, Kieu Dung Ly, Anne Milet and Yannick Vallée * Univ. Grenoble Alpes, CNRS, Département de Chimie Moléculaire, Campus, F-38058 Grenoble, France; [email protected] (I.S.); [email protected] (S.C.); [email protected] (K.D.L.); [email protected] (A.M.) * Correspondence: [email protected] Received: 28 September 2018; Accepted: 18 October 2018; Published: 19 October 2018 Abstract: The Strecker reaction of aldehydes with ammonia and hydrogen cyanide first leads to α-aminonitriles, which are then hydrolyzed to α-amino acids. However, before reacting with water, these aminonitriles can be trapped by aminothiols, such as cysteine or homocysteine, to give 5- or 6-membered ring heterocycles, which in turn are hydrolyzed to dipeptides. We propose that this two-step process enabled the formation of thiol-containing dipeptides in the primitive ocean. These small peptides are able to promote the formation of other peptide bonds and of heterocyclic molecules. Theoretical calculations support our experimental results. They predict that α-aminonitriles should be more reactive than other nitriles, and that imidazoles should be formed from transiently formed amidinonitriles. Overall, this set of reactions delineates a possible early stage of the development of organic chemistry, hence of life, on Earth dominated by nitriles and thiol-rich peptides (TRP). Keywords: origin of life; prebiotic chemistry; thiol-rich peptides; cysteine; aminonitriles; imidazoles 1. Introduction In ribosomes, peptide bonds are formed by the reaction of the amine group of an amino acid with an ester function. -

The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water
Orig Life Evol Biosph (2011) 41:399–412 DOI 10.1007/s11084-011-9243-4 PREBIOTIC CHEMISTRY The Relative Rates of Thiol–Thioester Exchange and Hydrolysis for Alkyl and Aryl Thioalkanoates in Water Paul J. Bracher & Phillip W. Snyder & Brooks R. Bohall & George M. Whitesides Received: 14 April 2011 /Accepted: 16 June 2011 / Published online: 5 July 2011 # Springer Science+Business Media B.V. 2011 Abstract This article reports rate constants for thiol–thioester exchange (kex), and for acid- mediated (ka), base-mediated (kb), and pH-independent (kw) hydrolysis of S-methyl thioacetate and S-phenyl 5-dimethylamino-5-oxo-thiopentanoate—model alkyl and aryl thioalkanoates, respectively—in water. Reactions such as thiol–thioester exchange or aminolysis could have generated molecular complexity on early Earth, but for thioesters to have played important roles in the origin of life, constructive reactions would have needed to compete effectively with hydrolysis under prebiotic conditions. Knowledge of the kinetics of competition between exchange and hydrolysis is also useful in the optimization of systems where exchange is used in applications such as self-assembly or reversible binding. For the alkyl thioester S-methyl thioacetate, which has been synthesized in −5 −1 −1 −1 −1 −1 simulated prebiotic hydrothermal vents, ka = 1.5×10 M s , kb = 1.6×10 M s , and −8 −1 kw = 3.6×10 s . At pH 7 and 23°C, the half-life for hydrolysis is 155 days. The second- order rate constant for thiol–thioester exchange between S-methyl thioacetate and 2- −1 −1 sulfonatoethanethiolate is kex = 1.7 M s . -

Identification of a Thioesterase Bottleneck in the Pikromycin Pathway Through Full-Module Processing of Unnatural Pentaketides
Article pubs.acs.org/JACS Identification of a Thioesterase Bottleneck in the Pikromycin Pathway through Full-Module Processing of Unnatural Pentaketides † ‡ † § † ‡ ⊥ ∥ Douglas A. Hansen, , Aaron A. Koch, , and David H. Sherman*, , , , † ‡ § ⊥ Life Sciences Institute, Department of Medicinal Chemistry, Cancer Biology Graduate Program, Department of Chemistry, and ∥ Department of Microbiology & Immunology, University of Michigan, Ann Arbor, Michigan 48109, United States *S Supporting Information ABSTRACT: Polyketide biosynthetic pathways have been engineered to generate natural product analogs for over two decades. However, manipulation of modular type I polyketide synthases (PKSs) to make unnatural metabolites commonly results in attenuated yields or entirely inactive pathways, and the mechanistic basis for compromised production is rarely elucidated since rate-limiting or inactive domain(s) remain unidentified. Accordingly, we synthesized and assayed a series of modified pikromycin (Pik) pentaketides that mimic early pathway engineering to probe the substrate tolerance of the PikAIII-TE module in vitro. Truncated pentaketides were processed with varying efficiencies to corresponding macrolactones, while pentaketides with epimerized chiral centers were poorly processed by PikAIII-TE and failed to generate 12-membered ring products. Isolation and identification of extended but prematurely offloaded shunt products suggested that the Pik thioesterase (TE) domain has limited substrate flexibility and functions as a gatekeeper in the processing of -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Organic Chemistry/Fourth Edition: E-Text
CHAPTER 17 ALDEHYDES AND KETONES: NUCLEOPHILIC ADDITION TO THE CARBONYL GROUP O X ldehydes and ketones contain an acyl group RC± bonded either to hydrogen or Ato another carbon. O O O X X X HCH RCH RCRЈ Formaldehyde Aldehyde Ketone Although the present chapter includes the usual collection of topics designed to acquaint us with a particular class of compounds, its central theme is a fundamental reaction type, nucleophilic addition to carbonyl groups. The principles of nucleophilic addition to alde- hydes and ketones developed here will be seen to have broad applicability in later chap- ters when transformations of various derivatives of carboxylic acids are discussed. 17.1 NOMENCLATURE O X The longest continuous chain that contains the ±CH group provides the base name for aldehydes. The -e ending of the corresponding alkane name is replaced by -al, and sub- stituents are specified in the usual way. It is not necessary to specify the location of O X the ±CH group in the name, since the chain must be numbered by starting with this group as C-1. The suffix -dial is added to the appropriate alkane name when the com- pound contains two aldehyde functions.* * The -e ending of an alkane name is dropped before a suffix beginning with a vowel (-al) and retained be- fore one beginning with a consonant (-dial). 654 Back Forward Main Menu TOC Study Guide TOC Student OLC MHHE Website 17.1 Nomenclature 655 CH3 O O O O CH3CCH2CH2CH CH2 CHCH2CH2CH2CH HCCHCH CH3 4,4-Dimethylpentanal 5-Hexenal 2-Phenylpropanedial When a formyl group (±CHœO) is attached to a ring, the ring name is followed by the suffix -carbaldehyde. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Variations in the Structure and Reactivity of Thioester Functionalized Self-Assembled Monolayers and Their Use for Controlled Surface Modification
Variations in the structure and reactivity of thioester functionalized self-assembled monolayers and their use for controlled surface modification Inbal Aped, Yacov Mazuz and Chaim N. Sukenik* Full Research Paper Open Access Address: Beilstein J. Nanotechnol. 2012, 3, 213–220. Department of Chemistry and Institute for Nanotechnology and doi:10.3762/bjnano.3.24 Advanced Materials, Bar-Ilan University, Ramat-Gan, Israel 52900 Received: 01 December 2011 Email: Accepted: 10 February 2012 Chaim N. Sukenik* - [email protected] Published: 09 March 2012 * Corresponding author This article is part of the Thematic Series "Self-assembly at solid surfaces". Keywords: siloxane-anchored self-assembled monolayers; sulfonated interfaces; Guest Editors: S. R. Cohen and J. Sagiv surface chemistry © 2012 Aped et al; licensee Beilstein-Institut. License and terms: see end of document. Abstract Thioester-functionalized, siloxane-anchored, self-assembled monolayers provide a powerful tool for controlling the chemical and physical properties of surfaces. The thioester moiety is relatively stable to long-term storage and its structure can be systematically varied so as to provide a well-defined range of reactivity and wetting properties. The oxidation of thioesters with different-chain- length acyl groups allows for very hydrophobic surfaces to be transformed into very hydrophilic, sulfonic acid-bearing, surfaces. Systematic variation in the length of the polymethylene chain has also allowed us to examine how imbedding reaction sites at various depths in a densely packed monolayer changes their reactivity. π-Systems (benzene and thiophene) conjugated to the thioester carbonyl enable the facile creation of photoreactive surfaces that are able to use light of different wavelengths. -

215-216 HH W12-Notes-Ch 15
Chem 215 F12 Notes Notes – Dr. Masato Koreeda - Page 1 of 17. Date: October 5, 2012 Chapter 15: Carboxylic Acids and Their Derivatives and 21.3 B, C/21.5 A “Acyl-Transfer Reactions” I. Introduction Examples: note: R could be "H" R Z R O H R O R' ester O carboxylic acid O O an acyl group bonded to R X R S acid halide* R' an electronegative atom (Z) thioester O X = halogen O R' R, R', R": alkyl, alkenyl, alkynyl, R O R' R N or aryl group R" amide O O O acid anhydride one of or both of R' and R" * acid halides could be "H" R F R Cl R Br R I O O O O acid fluoride acid chloride acid bromide acid iodide R Z sp2 hybridized; trigonal planar making it relatively "uncrowded" O The electronegative O atom polarizes the C=O group, making the C=O carbon "electrophilic." Resonance contribution by Z δ * R Z R Z R Z R Z C C C C O O O δ O hybrid structure The basicity and size of Z determine how much this resonance structure contributes to the hybrid. * The more basic Z is, the more it donates its electron pair, and the more resonance structure * contributes to the hybrid. similar basicity O R' Cl OH OR' NR'R" Trends in basicity: O weakest increasing basiciy strongest base base Check the pKa values of the conjugate acids of these bases. Chem 215 F12 Notes Notes –Dr. Masato Koreeda - Page 2 of 17. -
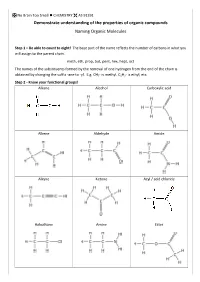
IUPAC Naming
No Brain Too Small CHEMISTRY AS 91391 Demonstrate understanding of the properties of organic compounds Naming Organic Molecules Step 1 – Be able to count to eight! The base part of the name reflects the number of carbons in what you will assign to the parent chain. meth, eth, prop, but, pent, hex, hept, oct The names of the substituents formed by the removal of one hydrogen from the end of the chain is obtained by changing the suffix -ane to -yl. E.g. CH3- is methyl, C2H5- is ethyl, etc. Step 2 - Know your functional groups! Alkane Alcohol Carboxylic acid Alkene Aldehyde Amide Alkyne Ketone Acyl / acid chloride Haloalkane Amine Ester No Brain Too Small CHEMISTRY AS 91391 This is NOT an exhaustive list of rules but a guide for L3 NCEA. Identify the principle functional group in the structure. If there is only ONE functional group then this is the principle functional group e.g. CH3CH2CH2COOH will have the suffix -oic acid as the carboxylic acid is the principle functional group. In cases where compounds have more than one functional group, then the principle functional group is decided by a priority order. carboxylic acids > acid derivatives* > aldehydes > ketones > alcohols > amines *esters, acyl/acid chlorides and amides 3-hydroxybutanoic acid 4-aminopentanoic acid as the carboxylic acid functional group takes priority Fluoro-, chloro-, bromo-, iodo- and alkyl groups have “no priority”. Their numbering is governed by the lowest sum rule. 2,3-dichloro-4-methylhexane and NOT 4,5-dichloro-2-methylhexane (as 2+3+4 = 9 whereas 2+4+5 = 11) No Brain Too Small CHEMISTRY AS 91391 Rules 1.