Aberrant Intracellular Calcium Cycling in the Heart
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Aquaporin Channels in the Heart—Physiology and Pathophysiology
International Journal of Molecular Sciences Review Aquaporin Channels in the Heart—Physiology and Pathophysiology Arie O. Verkerk 1,2,* , Elisabeth M. Lodder 2 and Ronald Wilders 1 1 Department of Medical Biology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] 2 Department of Experimental Cardiology, Amsterdam University Medical Centers, University of Amsterdam, 1105 AZ Amsterdam, The Netherlands; [email protected] * Correspondence: [email protected]; Tel.: +31-20-5664670 Received: 29 March 2019; Accepted: 23 April 2019; Published: 25 April 2019 Abstract: Mammalian aquaporins (AQPs) are transmembrane channels expressed in a large variety of cells and tissues throughout the body. They are known as water channels, but they also facilitate the transport of small solutes, gasses, and monovalent cations. To date, 13 different AQPs, encoded by the genes AQP0–AQP12, have been identified in mammals, which regulate various important biological functions in kidney, brain, lung, digestive system, eye, and skin. Consequently, dysfunction of AQPs is involved in a wide variety of disorders. AQPs are also present in the heart, even with a specific distribution pattern in cardiomyocytes, but whether their presence is essential for proper (electro)physiological cardiac function has not intensively been studied. This review summarizes recent findings and highlights the involvement of AQPs in normal and pathological cardiac function. We conclude that AQPs are at least implicated in proper cardiac water homeostasis and energy balance as well as heart failure and arsenic cardiotoxicity. However, this review also demonstrates that many effects of cardiac AQPs, especially on excitation-contraction coupling processes, are virtually unexplored. -

Supplemental Material
Supplemental Table B ARGs in alphabetical order Symbol Title 3 months 6 months 9 months 12 months 23 months ANOVA Direction Category 38597 septin 2 1557 ± 44 1555 ± 44 1579 ± 56 1655 ± 26 1691 ± 31 0.05219 up Intermediate 0610031j06rik kidney predominant protein NCU-G1 491 ± 6 504 ± 14 503 ± 11 527 ± 13 534 ± 12 0.04747 up Early Adult 1G5 vesicle-associated calmodulin-binding protein 662 ± 23 675 ± 17 629 ± 16 617 ± 20 583 ± 26 0.03129 down Intermediate A2m alpha-2-macroglobulin 262 ± 7 272 ± 8 244 ± 6 290 ± 7 353 ± 16 0.00000 up Midlife Aadat aminoadipate aminotransferase (synonym Kat2) 180 ± 5 201 ± 12 223 ± 7 244 ± 14 275 ± 7 0.00000 up Early Adult Abca2 ATP-binding cassette, sub-family A (ABC1), member 2 958 ± 28 1052 ± 58 1086 ± 36 1071 ± 44 1141 ± 41 0.05371 up Early Adult Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A 136 ± 8 147 ± 6 147 ± 13 155 ± 9 185 ± 13 0.01272 up Midlife Acadl acetyl-Coenzyme A dehydrogenase, long-chain 423 ± 7 456 ± 11 478 ± 14 486 ± 13 512 ± 11 0.00003 up Early Adult Acadvl acyl-Coenzyme A dehydrogenase, very long chain 426 ± 14 414 ± 10 404 ± 13 411 ± 15 461 ± 10 0.01017 up Late Accn1 amiloride-sensitive cation channel 1, neuronal (degenerin) 242 ± 10 250 ± 9 237 ± 11 247 ± 14 212 ± 8 0.04972 down Late Actb actin, beta 12965 ± 310 13382 ± 170 13145 ± 273 13739 ± 303 14187 ± 269 0.01195 up Midlife Acvrinp1 activin receptor interacting protein 1 304 ± 18 285 ± 21 274 ± 13 297 ± 21 341 ± 14 0.03610 up Late Adk adenosine kinase 1828 ± 43 1920 ± 38 1922 ± 22 2048 ± 30 1949 ± 44 0.00797 up Early -
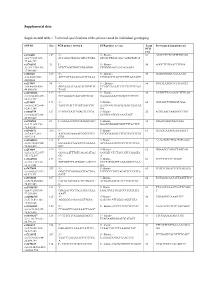
Supplemental Data Supplemental Table 1. Technical Specifications of the Primers Used for Individual Genotyping
Supplemental data Supplemental table 1. Technical specifications of the primers used for individual genotyping SNP ID Size PCR primer forward PCR primer reverse Temp Pyrosequencing primer(s) PCR (°C) rs776108 127 5’- 5’- Biotin- 61 ATACCTCTCATTTTGCAG chr3:77,825,927- ACCAGGCTAGGCATGCTATA GTCACTTAACAGCAGTGTGTCA 77,826,729 rs3746192 92 5’- 5’- Biotin- 56 AGGCTTGTAACCTGGA chr19:17,946,368- ATTCTAGGTGGCATGAGGG CTGGGGAGCAACAGAAGA 17,946,488 rs988147 169 5' - 5' - Biotine- 53 GCAGGGGGTAGAAATG chr6:45282108- AGCCATTAAAGAATTTCAAA TTGGATTTTATTCTTGTAATAGG 45282608 rs227849 94 5' - 5' - Biotine- 56 GGTTTAAGGTCTTTGCAT chr6:44,806,436- AGGAAAATAAACATGTGGTT TCTACCAATATTTTCTTTCGTAG 44,806,936 TAAG T rs10733833 127 5' - 5' - Biotine- 58 CATGTTTAAAACCTTTCAG chr10:68,418,227- GCCAAAACCAACAGTTCAT GAAAAAAATTGCACCTGTCTC 68,418,727 rs322609 197 5’- 5’-Biotin- 60 GGTAGCTGTGGGTGGA chr16:62,432,604- TAGTTGATTTTGCCAACCTG AAATGGGTGACAGAAGTAATAA 62,433,104 GA rs1884779 127 5’-TGGCTATTGGAGTTCTCA 5’-Biotin- 55 AGTGAATTAAGGGCTTGT chr20:45,857,969- CCATCCATCCCAAATAGT 45,858,469 rs4703908 83 5’- GAAAATGCCCAAGGTGAC 5’-Biotin- 52 GTGACAGTGGGCAAA chr5:71,802,353- GAATGTGGGTGTGTTTTACTCT 71,802,853 rs6946871 290 5’- 5’-Biotin- 61 GGAGGAAAGGAAAAGTT chr7:4,037,310- ATCAGATAAAATCGGCTTCT TCGGGAAGGTTTTTGTACTTTTG 4,037,810 GTG rs11865033 123 5’- 5’-Biotin- 61 AAAGTCTCTTCCTATGAGC chr16:78,082,945- AATAAACCAAGCCCTGAAAA ACTAAAATCCCCCTTTCCTCCA 78,083,445 GTC rs247004 170 5’- 5’-Biotin- 62 GGAAGCCAGACTAGCAG chr5:131,372,007- GGGGAATTTGTCAGAGATAG GGGATCCTCTACCATCCAAATA 131,372,507 GG -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Disease Mutations in the Ryanodine Receptor N-Terminal Region Couple to a Mobile Intersubunit Interface
ARTICLE Received 1 Oct 2012 | Accepted 15 Jan 2013 | Published 19 Feb 2013 DOI: 10.1038/ncomms2501 OPEN Disease mutations in the ryanodine receptor N-terminal region couple to a mobile intersubunit interface Lynn Kimlicka1, Kelvin Lau1, Ching-Chieh Tung1 & Filip Van Petegem1 Ryanodine receptors are large channels that release Ca2 þ from the endoplasmic and sar- coplasmic reticulum. Hundreds of RyR mutations can cause cardiac and skeletal muscle disorders, yet detailed mechanisms explaining their effects have been lacking. Here we compare pseudo-atomic models and propose that channel opening coincides with widen- ing of a cytoplasmic vestibule formed by the N-terminal region, thus altering an interface targeted by 20 disease mutations. We solve crystal structures of several disease mutants that affect intrasubunit domain–domain interfaces. Mutations affecting intrasubunit ionic pairs alter relative domain orientations, and thus couple to surrounding interfaces. Buried disease mutations cause structural changes that also connect to the intersubunit contact area. These results suggest that the intersubunit contact region between N-terminal domains is a prime target for disease mutations, direct or indirect, and we present a model whereby ryanodine receptors and inositol-1,4,5-trisphosphate receptors are activated by altering domain arrangements in the N-terminal region. 1 Department of Biochemistry and Molecular Biology, Life Sciences Institute, University of British Columbia, Vancouver, British Columbia, Canada V6T 1Z3. Correspondence and requests for materials should be addressed to F.V.P. (email: fi[email protected]). NATURE COMMUNICATIONS | 4:1506 | DOI: 10.1038/ncomms2501 | www.nature.com/naturecommunications 1 & 2013 Macmillan Publishers Limited. All rights reserved. -

Physiological and Pathophysiological Regulation of the Ryanodine Receptor in Skeletal Muscle
Physiological and pathophysiological regulation of the ryanodine receptor in skeletal muscle Alisa Umanskaya Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2015 © 2015 Alisa Umanskaya All rights reserved Abstract Physiological and pathophysiological regulation of ryanodine receptor in skeletal muscle Alisa Umanskaya Ryanodine receptor calcium release channels are essential for skeletal muscle contraction, as they mediate the release of calcium ions from intracellular stores into the cytosol. The data presented in this dissertation demonstrate the evolutionarily conserved mechanisms of skeletal muscle ryanodine receptor regulation in the physiological and pathophysiological states. Adrenergic stimulation causes increased skeletal muscle force, however, despite the well- established role of this physiological response, the molecular mechanism is not known. Here we present a mechanism whereby phosphorylation of a single amino acid on the ryanodine receptor is a key signal in the physiological stress-induced inotropic response in mouse skeletal muscle. Therefore acute post-translational modifications of ryanodine receptor channels are important for healthy muscle contraction. Conversely, chronic stress-induced post-translational modifications result in poorly functioning murine ryanodine receptor channels that contribute to skeletal muscle dysfunction in age- dependent skeletal muscle weakness and Muscular Dystrophies. Finally, we present data that demonstrates striking evolutionary conservation in ryanodine receptor regulation in the physiological and pathophysiological states between mice and C. elegans. This work has broad implications for understanding the underlying mechanisms of skeletal muscle contraction and important disorders that affect human health. Furthermore, this works presents ryanodine receptor channels as a viable therapeutic target for age-related skeletal muscle weakness, Muscular Dystrophies, and also implicates C. -

2006 Annual Report, My First As Improving Links with and Transfer of Information to and from Our Partners, Director
ANNUAL REPORT 2006 The Menzies School of Health Research (MSHR) was established in 1985 as a body corporate of the Northern Territory (NT) Government under the Menzies School of Health Research Act 1985 (The MSHR Act). This act was amended in 2004 to formalise the relationship with Charles Darwin University (CDU). MSHR is now a controlled-entity of CDU and constitutes a school within the university’s Institute of Advanced Studies. Timika Dili Darwin MSHR Darwin Headquarters Alice Springs Menzies staff and facilities are uniquely positioned to conduct We also operate a smaller unit in Alice Springs co-located with active research across the country’s tropical north, throughout the Centre for Remote Health, and a joint research facility with remote Indigenous communities, and in partnership with our the Indonesian Ministry of Health’s National Institute of Health neighbouring countries to the north. Research and Development, in Timika, Indonesia. MSHR headquarters are located on the Royal Darwin Hospital Campus, providing office accommodation for the majority of our In the spirit of respect, Menzies School of Health Research staff and students. It also houses a well equipped and highly acknowledges the people and elders of the Aboriginal and regarded laboratory with (PC2 and PC3 containment facilities), Torres Strait Islander Nations, who are the Traditional Owners conducting leading edge science ranging from analysis of of the land and seas of Australia. snake venom, soil samples for Melioidosis, scabies mite drug resistance to pathogenicity of streptococci. For the purpose of this document ‘Indigenous’ refers to Australia’s Aboriginal and Torres Strait Islander peoples. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

RYR2 Gene Ryanodine Receptor 2
RYR2 gene ryanodine receptor 2 Normal Function The RYR2 gene provides instructions for making a protein called ryanodine receptor 2. This protein is part of a family of ryanodine receptors, which form channels that transport positively charged calcium atoms (calcium ions) within cells. Channels made with the ryanodine receptor 2 protein are found in heart (cardiac) muscle cells called myocytes. These channels are embedded in the outer membrane of a cell structure called the sarcoplasmic reticulum, which acts as a storage center for calcium ions. The RYR2 channel controls the flow of calcium ions out of the sarcoplasmic reticulum. For the heart to beat normally, the cardiac muscle must tense (contract) and relax in a coordinated way. This cycle of muscle contraction and relaxation results from the precise control of calcium ions within myocytes. In response to certain signals, the RYR2 channel releases calcium ions from the sarcoplasmic reticulum into the surrounding cell fluid (the cytoplasm). The resulting increase in calcium ion concentration triggers the cardiac muscle to contract, which pumps blood out of the heart. Calcium ions are then transported back into the sarcoplasmic reticulum, and the cardiac muscle relaxes. In this way, the release and reuptake of calcium ions in myocytes produces a regular heart rhythm. Health Conditions Related to Genetic Changes Catecholaminergic polymorphic ventricular tachycardia More than 200 mutations in the RYR2 gene have been found to cause catecholaminergic polymorphic ventricular tachycardia (CPVT), a heart condition characterized by an abnormal heart rhythm (arrhythmia) during exercise or emotional stress, which can be fatal. Almost all of the RYR2 gene mutations involved in CPVT change single protein building blocks (amino acids) in the ryanodine receptor 2 protein. -

Investigation Into the Cardiotoxic Effects of B- Adrenergic Receptor Agonists in Myocardial Ischaemia/Reperfusion Injury
Investigation into the cardiotoxic effects of b- adrenergic receptor agonists in myocardial ischaemia/reperfusion injury Nagra, A. S. Submitted version deposited in Coventry University’s Institutional Repository Original citation: Nagra, A. S. (2016) Investigation into the cardiotoxic effects of b- adrenergic receptor agonists in myocardial ischaemia/reperfusion injury. Unpublished PhD Thesis. Coventry: Coventry University Copyright © and Moral Rights are retained by the author. A copy can be downloaded for personal non-commercial research or study, without prior permission or charge. This item cannot be reproduced or quoted extensively from without first obtaining permission in writing from the copyright holder(s). The content must not be changed in any way or sold commercially in any format or medium without the formal permission of the copyright holders. Some materials have been removed from this thesis due to third party copyright. Pages where material has been removed are clearly marked in the electronic version. The unabridged version of the thesis can be viewed at the Lanchester Library, Coventry University. Investigation into the cardiotoxic effects of b- adrenergic receptor agonists in myocardial ischaemia/reperfusion injury By Aarondeep Singh Nagra January 2016 Supervisory team: Dr. Afthab Hussain, Professor Helen Maddock & Dr. Christopher Mee A thesis submitted in partial fulfilment of the University’s requirements for the Degree of Doctor of Philosophy Acknowledgements I would like to give thanks to my supervisory team Dr. Afthab Hussain, Professor Helen Maddock and Dr. Christopher Mee, whose guidance throughout has been invaluable allowing me to confidently go on to complete my thesis. I would also like to give special thanks to my colleagues; Mayel Gharanaei, Jawad Khan, Katherine Harvey and Maryam Babba for making our time in the lab together a period for us all to look back on and reminisce the good times. -

AMREP Research Report 2011
The Alfred Medical Research and Education Precinct Research Report 2011 Research and Education Precinct Medical Research The Alfred 2 011 The Alfred Medical Research & Education Precinct Alfred Medical Research and Education Precinct Commercial Road, Melbourne, Victoria 3004, Australia www.amrep.org.au The Alfred Medical Research and Education Precinct The Alfred Medical Research and Education Precinct - AMREP - is a partnership between Alfred Health, Monash University, Baker IDI Heart and Diabetes Institute, Burnet Institute, La Trobe University and Deakin University. AMREP is located on the campus of The Alfred hospital, Melbourne. Alfred Medical Research & Education Precinct Commercial Road Melbourne, Victoria 3004 Australia www.amrep.org.au Acknowledgements Produced by Research Office, Alfred Health and Baker IDI Heart and Diabetes Institute Design by abCreative | abCreative.com CONTENTS AMREP Highlights 2011/2012 2 Andrew Way Research Output 4 Chief Executive, Human Ethics 6 Alfred Health Animal Ethics 7 Chair, AMREP Council Baker IDI Heart and Diabetes Institute 8 Nucleus Network 14 Burnet Institute 16 As I set out in my 2010/11 report, the benefits of Academic Health Monash School of Public Health and Preventive Medicine 24 Science Centres (AHSCs) continue to be widely discussed. Both Epidemiology and Preventive Medicine 25 State and Federal government departments are known to be Global Health 27 taking an increasing interest in their potential. AMREP, established Centre for Obesity Research and Education 28 in 2002, is Australia’s first and longest existing example of such Australasian Cochrane Centre 29 an endeavour, although when created no one at the time would Rheumatology (Musculoskeletal Epidemiology) 30 have thought – Academic Health Science Centre.