Methods for the Determination of Chlorine, Hydrogen-Sulfide, and Hydrogen-Cyanide Utilizing Permeation Sampling
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SAFETY DATA SHEET Nonflammable Gas Mixture: Cyanogen Chloride 1-999Ppm / Nitrogen 99%
SAFETY DATA SHEET Nonflammable Gas Mixture: Cyanogen Chloride 1-999ppm / Nitrogen 99% Section 1. Identification GHS product identifier : Nonflammable Gas Mixture: Cyanogen Chloride 1-999ppm / Nitrogen 99% Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 012226 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Compressed gas substance or mixture GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Prevention : Not applicable. Response : Not applicable. Storage : Protect from sunlight when ambient temperature exceeds 52°C/125°F. Store in a well- ventilated place. Disposal : Not applicable. Hazards not otherwise : In addition to any other important health or physical hazards, this product may displace classified oxygen and cause rapid suffocation. Section 3. Composition/information on ingredients Substance/mixture : Mixture Other means of : Not available. -

Abbreviations of Chemical Warfare Agents, 152 Acetylcholinesterase
Index Abbreviations of chemical warfare agents, Blood ChE inhibition, nerve agent critical 152 toxic effect on, 23 Acetylcholinesterase (AChE) inhibition, Blood ChE inhibition, nerve agent species nerve agents and, 18 variation of, 21 Acetylthiocholine iodide, RBC-ChE activ Blood cholinesterases, nerve agent effects ity and, 22 on, 20 AChE (acetylcholinesterase) inhibition, Butyrylcholinesterase (plasma-ChE), nerve agents and, 18 nerve agent effects on, 20 Acronyms for chemical warfare agents, 152 Acute inhalation toxicity, CK and, 114 Carboxylesterases (aliesterases), nerve Acute toxicity, CK (cyanogen chloride) agent effects on, 21 and, 113 Carcinogenicity, CK and, 122 Acute toxicity, GA and, 68 Carcinogenicity, GA and, 73 Acute toxicity, GB and, 78 Carcinogenicity, GB and, 90 Acute toxicity, GD and, 94 Carcinogenicity, HD and, 38 Acute toxicity, HD and, 30 Carcinogenicity, HN2 and, 53 Acute toxicity, HN2 and, 51 Carcinogenicity, HT and, 49 Acute toxicity, HT and, 49 Carcinogenicity, lewisite and, 107 Acute toxicity, lewisite and, 103 Carcinogenicity, VX and, 61 Acute toxicity, T sulfur mustard and, 50 ChE (cholinesterase), nerve agent effects Acute toxicity, VX and, 54 on, 20 Agent CK, see CK or cyanogen chloride ChE inhibition, nerve agent potency on, Agent GA, see GA or Tabun 23 Agent GB, see GB or Sarin Chemical warfare agent disposal tech Agent GD, see GD or Soman niques,3 Agent HD, see HD or Sulfur mustard HD Chemical warfare agent non-stockpile Agent HN2, see HN2 or Nitrogen mus- sites U.S., 4 tard Chemical warfare agents, 1 ff. Agent HT, see HT or Sulfur mustard HT Chemical warfare agents, acronyms/abbre Agent L, see L or Lewsite viations of, 152 Agent T, see T Sulfur mustard Chemical warfare agents, air exposure lim Agent VX, see VX its of, 7 Aliesterases (carboxylesterases), nerve Chemical warfare agents, environmental agent effects on, 21 cleanup of, 3 Anticholinesterase agents, 18 Chemical warfare agents, environmental Army chemical destruction, 3 ff. -

Threshold Limit Values (TLV) and Immediately Dangerous to Life and Health (IDLH) Values
Reference Information 442 Threshold Limit Values (TLV) and Immediately Dangerous to Life and Health (IDLH) values Matheson Tri-Gas assumes no responsibility for the accuracy of the TLV TLV information listed. As TLV and IDLH values may change, the current Substance TWA STEL IDLH guidelines from the following sources should be consulted for up-to-date accuracy. Fluorine 1 2 25 Formaldehyde - 0.3C 20 Sources: TLV-TWA and TLV-STEL data extracted from the 2005 Threshold Limit Values & Biological Exposure Indices, copyright 2005 by the American Conference of Formic Acid 5 10 30 Governmental Industrial Hygienists (ACGIH). IDLH values extracted from the Furfural 2 - 100 NIOSH Pocket Guide to Chemical Hazards, 2004 published by the National Institute Gasoline 300 500 - for Occupational Safety and Health (NIOSH). Germanium tetrahydride 0.2 - - Note: All concentrations in parts per million (ppm) unless otherwise noted. Glutaraldehyde - 0.05C - “C” indicates Ceiling Limit. Heptane 400 500 750 TLV TLV n-Hexane 50 - 1,100 Substance TWA STEL IDLH Hydrazine 0.01 - 50 Acetaldehyde - 25C 2,000 Hydrogen Bromide - 2C 30 Acetic Acid 10 15 50 Hydrogen Chloride - 2C 50 Acetone 500 750 2,500 Hydrogen Cyanide - 4.7C 50 Acetonitirle 20 - 500 Hydrogen Fluoride 0.5 2C 30 Acrolein - 0.1C 2 Hydrogen Peroxide 1 - 75 Acrylonitrile 2 - 85 Hydrogen Selenide 0.05 - 1 Ammonia 25 35 300 Hydrogen Sulfide 10 15 100 Arsine 0.05 - 3 Iodine - 0.1C 2 Benzene 0.5 2.5 500 Isopropyl Alcohol 200 400 2,000 Boron Trifluoride - 1C 25 Methyl Alcohol 200 250 6,000 Bromine 0.1 0.2 3 Methylamine -

NIOSH Guidance -Filtration and Air Cleaning Systems to Protect Building
Guidance for Filtration and Air-Cleaning Systems to Protect Building Environments from Airborne Chemical, Biological, or Radiological Attacks Department of Health and Human Services Centers for Disease Control and Prevention National Institute for Occupational Safety and Health April 2003 ii Ordering Information To receive documents or other information about occupational safety and health topics, contact the National Institute for Occupational Safety and Health (NIOSH) at NIOSH Publications Dissemination 4676 Columbia Parkway Cincinnati, OH 45226-1998 Telephone: 1-800-35-NIOSH (1-800-356-4674) Fax: 1-513-533-8573 E-mail: [email protected] or visit the NIOSH Web site at www.cdc.gov/niosh This document is in the public domain and may be freely copied or reprinted. Disclaimer: Mention of any company, product, policy, or the inclusion of any reference does not constitute endorse- ment by NIOSH. DHHS (NIOSH) Publication No. 2003-136 iii Foreword The Occupational Safety and Health Act of 1970 [Public Law 91-596] assures so far as possible every working man and woman in the Nation safe and healthful working conditions. The Act charges the National Institute for Occupational Safety and Health (NIOSH) with conducting research and making science-based recommenda- tions to prevent work-related illness, injury, disability, and death. On October 8, 2001, the President of the United States established by executive order the Office of Homeland Security (OHS), which is mandated “to develop and coordinate the implementation of a com- prehensive national strategy to secure the United States from ter- rorist threats or attacks.” In January 2002, the OHS formed the Interagency Workgroup on Building Air Protection under the Medical and Public Health Preparedness Policy Coordinating Committee of the OHS. -

Chemical Weapons Technology Section 4—Chemical Weapons Technology
SECTION IV CHEMICAL WEAPONS TECHNOLOGY SECTION 4—CHEMICAL WEAPONS TECHNOLOGY Scope Highlights 4.1 Chemical Material Production ........................................................II-4-8 4.2 Dissemination, Dispersion, and Weapons Testing ..........................II-4-22 • Chemical weapons (CW) are relatively inexpensive to produce. 4.3 Detection, Warning, and Identification...........................................II-4-27 • CW can affect opposing forces without damaging infrastructure. 4.4 Chemical Defense Systems ............................................................II-4-34 • CW can be psychologically devastating. • Blister agents create casualties requiring attention and inhibiting BACKGROUND force efficiency. • Defensive measures can be taken to negate the effect of CW. Chemical weapons are defined as weapons using the toxic properties of chemi- • Donning of protective gear reduces combat efficiency of troops. cal substances rather than their explosive properties to produce physical or physiologi- • Key to employment is dissemination and dispersion of agents. cal effects on an enemy. Although instances of what might be styled as chemical weapons date to antiquity, much of the lore of chemical weapons as viewed today has • CW are highly susceptible to environmental effects (temperature, its origins in World War I. During that conflict “gas” (actually an aerosol or vapor) winds). was used effectively on numerous occasions by both sides to alter the outcome of • Offensive use of CW complicates command and control and battles. A significant number of battlefield casualties were sustained. The Geneva logistics problems. Protocol, prohibiting use of chemical weapons in warfare, was signed in 1925. Sev- eral nations, the United States included, signed with a reservation forswearing only the first use of the weapons and reserved the right to retaliate in kind if chemical weapons were used against them. -

TIH/PIH List
Hazardous Materials Designated as TIH/PIH (consolidated AAR and Railinc lists) 3/12/2007 STCC Proper Shipping Name 4921402 2-CHLOROETHANAL 4921495 2-METHYL-2-HEPTANETHIOL 4921741 3,5-DICHLORO-2,4,6-TRIFLUOROPYRIDINE 4921401 ACETONE CYANOHYDRIN, STABILIZED 4927007 ACROLEIN, STABILIZED 4921019 ALLYL ALCOHOL 4923113 ALLYL CHLOROFORMATE 4921004 ALLYLAMINE 4904211 AMMONIA SOLUTION 4920360 AMMONIA SOLUTIONS 4904209 AMMONIA, ANHYDROUS 4904210 AMMONIA, ANHYDROUS 4904879 AMMONIA, ANHYDROUS 4920359 AMMONIA, ANHYDROUS 4923209 ARSENIC TRICHLORIDE 4920135 ARSINE 4932010 BORON TRIBROMIDE 4920349 BORON TRICHLORIDE 4920522 BORON TRIFLUORIDE 4936110 BROMINE 4920715 BROMINE CHLORIDE 4918505 BROMINE PENTAFLUORIDE 4936106 BROMINE SOLUTIONS 4918507 BROMINE TRIFLUORIDE 4921727 BROMOACETONE 4920343 CARBON MONOXIDE AND HYDROGEN MIXTURE, COMPRESSED 4920399 CARBON MONOXIDE, COMPRESSED 4920511 CARBON MONOXIDE, REFRIGERATED LIQUID 4920559 CARBONYL FLUORIDE 4920351 CARBONYL SULFIDE 4920523 CHLORINE 4920189 CHLORINE PENTAFLUORIDE 4920352 CHLORINE TRIFLUORIDE 4921558 CHLOROACETONE, STABILIZED 4921009 CHLOROACETONITRILE 4923117 CHLOROACETYL CHLORIDE 4921414 CHLOROPICRIN 4920516 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920547 CHLOROPICRIN AND METHYL BROMIDE MIXTURES 4920392 CHLOROPICRIN AND METHYL CHLORIDE MIXTURES 4921746 CHLOROPIVALOYL CHLORIDE 4930204 CHLOROSULFONIC ACID 4920527 COAL GAS, COMPRESSED 4920102 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920303 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920304 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920305 COMMPRESSED GAS, TOXIC, FLAMMABLE, CORROSIVE, N.O.S. 4920101 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920300 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920301 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920324 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920331 COMPRESSED GAS, TOXIC, CORROSIVE, N.O.S. 4920165 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920378 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. 4920379 COMPRESSED GAS, TOXIC, FLAMMABLE, N.O.S. -
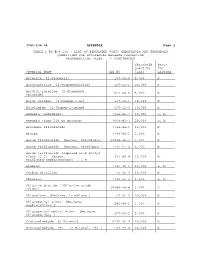
3745-104-04 APPENDIX Page 1 TABLE 1 to '68.130
3745-104-04 APPENDIX Page 1 TABLE 1 TO '68.130 - LIST OF REGULATED TOXIC SUBSTANCES AND THRESHOLD QUANTITIES FOR ACCIDENTAL RELEASE PREVENTION [ALPHABETICAL ORDER - 77 SUBSTANCES] Threshold Basis Quantity for Chemical Name CAS No (lbs) Listing Acrolein [2-Propenal] 107-02-8 5,000 b Acrylonitrile [2-Propenenitrile] 107-13-1 20,000 b Acrylyl chloride [2-Propenoyl 814-68-6 5,000 b chloride] Allyl alcohol [2-Propen-1-ol] 107-18-6 15,000 b Allylamine [2-Propen-1-amine] 107-11-9 10,000 b Ammonia (anhydrous) 7664-41-7 10,000 a, b Ammonia (conc 20% or greater) 7664-41-7 20,000 a, b Arsenous trichloride 7784-34-1 15,000 b Arsine 7784-42-1 1,000 b Boron trichloride [Borane, trichloro-] 10294-34-5 5,000 b Boron trifluoride [Borane, trifluoro-] 7637-07-2 5,000 b Boron trifluoride compound with methyl ether (1:1) [Boron, 353-42-4 15,000 b trifluoro[oxybis[metane]]-, T-4- Bromine 7726-95-6 10,000 a, b Carbon disulfide 75-15-0 20,000 b Chlorine 7782-50-5 2,500 a, b Chlorine dioxide [Chlorine oxide (ClO2)] 10049-04-4 1,000 c Chloroform [Methane, trichloro-] 67-66-3 20,000 b Chloromethyl ether [Methane, oxybis[chloro-] 542-88-1 1,000 b Chloromethyl methyl ether [Methane, chloromethoxy-] 107-30-2 5,000 b Crotonaldehyde [2-Butenal] 4170-30-3 20,000 b Crotonaldehyde, (E)- [2-Butenal, (E)-] 123-73-9 20,000 b 3745-104-04 Appendix 2 Threshold Basis Quantity for Chemical Name CAS No (lbs) Listing Cyanogen chloride 506-77-4 10,000 c Cyclohexylamine [Cyclohexanamine] 108-91-8 15,000 b Diborane 19287-45-7 2,500 b Dimethyldichlorosilane [Silane, dichlorodimethyl-] -

Hydrogen Cyanide and Cyanides: Human Health Aspects
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 61 HYDROGEN CYANIDE AND CYANIDES: HUMAN HEALTH ASPECTS Please note that the layout and pagination of this pdf file are not identical to the version in press First draft prepared by Prof. Fina Petrova Simeonova, Consultant, National Center of Hygiene, Medical Ecology and Nutrition, Sofia, Bulgaria; and Dr Lawrence Fishbein, Fairfax, Virginia, USA Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2004 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management -

Parameters for the Evaluation of the Fate, Transport, and Environmental Impacts of Chemical Agents in Marine Environments July 2007
Parameters for the Evaluation of the Fate, Transport, and Environmental Impacts of Chemical Agents in Marine Environments July 2007 3150 Fairview Park Drive South Falls Church, VA 22042-4519 Tel 703.610.2002 noblis.org 2007 Noblis, Inc. Company Confidential/ Parameters for the Evaluation of the Fate, Transport, and Environmental Impacts of Chemical Agents in Marine Environments July 2007 George O. Bizzigotti, Ph. D. Harry Castelly Ahmed M. Hafez, Ph. D. Wade H. B. Smith, Ph. D. Mark T. Whitmire Abstract 313 references were reviewed to obtain data relevant to the fate, transport, and environmental impacts of chemical agents in marine environments. The review covers the chemical agents phosgene, cyanogen chloride, hydrogen cyanide, sulfur mustard, Lewisite, nitrogen mustard (HN1), Tabun, Sarin, and VX. Parameters covered in the review are boiling point, melting point, density, vapor pressure, solubility, Henry’s Law constant, partition coefficients, dissociation constants, and hydrolysis rate constants and products. Recent ecotoxicity data are also discussed. Data gaps for these parameters and agents are identified and prioritized. In addition to the specific data gaps, the authors have identified several general issues that extend beyond single compounds or types of data. Given the available data and what they suggest will be the dominant fate and transport mechanisms, there are no data gaps that leave one unable to conduct a reasonable assessment for any agent. The primary source of uncertainty in the evaluation of the fate, transport, and environmental impacts of these agents remains the rate at which they are released into the environment. The data gaps identified in this work appear to cause a significantly smaller level of uncertainty. -

Chemical Warfare Agents
Agent Reference Common Name Looks/State Dispersion Odour Symptoms GA TABUN Colourless or Brown Liquid Liquid/Gas Fruity Pinpoint pupils, runny nose, drooling, coughing, GB SARIN Colourless Liquid Liquid/Gas Odourless tightness in chest, muscle twitching, nausea GD SOMAN Colourless Liquid Liquid/Gas Camphor (Mothballs) convulsions, coma, death. V Series Nerve Agents GF CYCLOSARIN Colourless Liquid Liquid/Gas Sweet & Musty `Novichok Series´ Nerve Substance 33, A230, A232, VX VX Colourless Liquid Liquid/Gas Odourless A234 etc., agents that were designed NOT to be VE (VX) Colourless Liquid Liquid/Gas Odourless detected by conventional Detection, Identification VG AMITON or TETRAM Colourless Liquid Liquid/Gas Odourless and Monitoring (DIM) equipment. VM EDEMO Colourless Liquid Liquid/Gas Odourless CG PHOSGENE Colourless Gas Yellow Gas Strong smell of Wood/Hay Coughing, eye, nose, throat Liquid irritation shortness of breath, frothy secretions CHEMICAL DP DIPHOSGENE Colourless Liquid Vapour/Liquid Strong smell of Wood a feeling of suffocating or drowning, death CL CHLORINE Yellow/Green Gas Gas Bleach AMMONIA/AZANE Colourless Gas Liquid/Gas Strong Urine smell Choking WARFARE DISULPHUR DECAFLUORIDE Colourless Gas Liquid/Gas PERFLUOROISOBUTENE Colourless Gas Liquid/Gas ACROLEIN/PROPENAL Colourless Liquid Acrid smell of Burned Fat AGENTS DIPHENYLCYANOARSINE CX PHOSGENE OXIME Solid Aerosol Unpleasant Watery and itchy eyes, runny nose, hoarseness ED ETHYLDICLOROARSINE Liquid Liquid/Aerosol Fruity or hacking cough, initial redness of skin, followed The following descriptions include odour. HD DISTILLED MUSTARD Colourless or Yellow Liquid Liquid/Gas Mustard/Garlic by blisters. HL MUSTARD-LEWISITE Liquid Liquid Mustard/Garlic Mustard has NO immediate DO NOT and NEVER intentionally smell/sniff effect. unknown chemicals. -

Fpm 8000 Anti-Terrorism Sensor for Chemical Warfare Agents
FPM 8000 ANTI-TERRORISM SENSOR FOR CHEMICAL WARFARE AGENTS The Control Equipment FPM 8000 Facility Protection Monitor is the product of 20 years of research and development in conjunction with Military and Homeland Security Authorities. FEATURES: • Detects 17 individual CWA and TIC THREAT Gases • Intelligent Nano Array Sensor (iNAS) technology • Long life highly stable sensor • Constant Sampling • Fast Response • Stand Alone Instrument • Wireless and wired outputs • Small form factor • 24V DC Powered APPLICATIONS: • Airports • Subways • Public Buildings • Shopping Malls • Government Facilities • Defense Facilities • Sensitive Infrastructures • Power Stations FPM 8000 ANTI-TERRORISM SENSOR FOR CHEMICAL WARFARE AGENTS GASES DETECTED & IDLH MEASURED: Acrolein 2ppm Lewisite 14ppb Acrylonitrile 85ppm Mustard 108ppb Ammonia 300ppm Nitrogen Dioxide 20ppm Chlorine 10ppm Phosgene 2ppm Cyanogen Chloride 20ppm Sarin 17ppb Ethylene Oxide 800ppm Sulphur Dioxide 100ppm Formaldehyde 20ppm Tabun 14ppb Hydrogen Chloride 50ppm VX 3ppb Hydrogen Cyanide 50ppm The FPM 8000 is based on revolutionary highly sensitive nano array technology which is at the heart of the intelligent sensor, making it capable of discriminating between gases and identifying target Chemical Warfare Agent gases (CWAs) and Toxic Industrial Compound gases (TICs). It senses 17 predominant THREAT gases used for terrorist purposes (5 CWAs and 12 TICs). Each THREAT gas is identified and its concentration is measured by the Intelligent Nano Array Sensor (iNAS). The FPM 8000 can be relied upon to alarm only when a credible threat is detected. The ( i NAS ) technology guards against false alarms, so common in instruments using other sensor technologies. The FPM 8000 utilizes industry standard RS 485 protocol to output data quantifying each of the 17 THREAT gas levels. -

Cyanogen Agents
J R Army Med Corps 2002; 148: 383-386 J R Army Med Corps: first published as 10.1136/jramc-148-04-08 on 1 December 2002. Downloaded from Cyanogen Agents GENERAL HCN + Na2S2O3 ‘HSCN + Na2SO3 This reaction with sodium thiosulphate Introduction forms the basis of a therapy of cyanide Cyanogen agents produce their effects by poisoning. Metal ions easily form complexes interfering with oxygen utilization at the with cyanide ions for example: cellular level. Their toxicity is primarily derived from the liberation of the CN-group. - 2- - Inhalation is the usual route of entry. The CoCl2 + 4CN ‘Co(CN) 4+ 2Cl . term "blood agents" has, in the past been These types of reaction are employed in used to describe cyanogen agents and is still reactive carbons where the charcoal is in widespread use. It should be noted, impregnated with metal ions in order to however, that not all "blood agents" are increase the absorptive capacity of charcoal. cyanogens (e.g. carbon monoxide). Similar reactions with complexed metal ions The commodity industrial chemical are also utilised in some forms of therapy. hydrogen cyanide (HCN (AC)) was used during WWI and is still attractive as an Detection improvised weapons fill because of its Automatic detectors are available which availability. Hydrogen cyanide is released detect attack concentrations of cyanide when cyanides (e.g. acetone cyanohydrine, vapour. DraegerTM tubes are also available, as sodium cyanide, potassium cyanide) are are water testing kits. spilled in water or subjected to acid. The cyanogen halides, cyanogen chloride (ClCN Protection (CK)) and cyanogen bromide (BrCN), were Hydrogen cyanide, because of its volatility used during WWI.