Identification of a Potential Mitotic Function for the Mammalian Nup50
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Persistent Transcription-Blocking DNA Lesions Trigger Somatic Growth Attenuation Associated with Longevity
ARTICLES Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity George A. Garinis1,2, Lieneke M. Uittenboogaard1, Heike Stachelscheid3,4, Maria Fousteri5, Wilfred van Ijcken6, Timo M. Breit7, Harry van Steeg8, Leon H. F. Mullenders5, Gijsbertus T. J. van der Horst1, Jens C. Brüning4,9, Carien M. Niessen3,9,10, Jan H. J. Hoeijmakers1 and Björn Schumacher1,9,11 The accumulation of stochastic DNA damage throughout an organism’s lifespan is thought to contribute to ageing. Conversely, ageing seems to be phenotypically reproducible and regulated through genetic pathways such as the insulin-like growth factor-1 (IGF-1) and growth hormone (GH) receptors, which are central mediators of the somatic growth axis. Here we report that persistent DNA damage in primary cells from mice elicits changes in global gene expression similar to those occurring in various organs of naturally aged animals. We show that, as in ageing animals, the expression of IGF-1 receptor and GH receptor is attenuated, resulting in cellular resistance to IGF-1. This cell-autonomous attenuation is specifically induced by persistent lesions leading to stalling of RNA polymerase II in proliferating, quiescent and terminally differentiated cells; it is exacerbated and prolonged in cells from progeroid mice and confers resistance to oxidative stress. Our findings suggest that the accumulation of DNA damage in transcribed genes in most if not all tissues contributes to the ageing-associated shift from growth to somatic maintenance that triggers stress resistance and is thought to promote longevity. Ageing represents the progressive functional decline that is exempted levels as a result of pituitary dysfunction (Snell and Ames mice) — have an from evolutionary selection because it largely occurs after reproduc- extended lifespan17–20. -

Identification of Proteins Interacting with Dysferlin Using the Tandem Affinity Purification Method
The Open Cell Development & Biology Journal, 2008, 1, 17-23 17 Open Access Identification of Proteins Interacting with Dysferlin Using the Tandem Affinity Purification Method Maziar Assadi*,1, Thomas Schindler1, Bernd Muller1, John D. Porter2, Markus A. Ruegg3 and Hanno Langen1 1F. Hoffmann-La Roche, Roche Center for Medical Genomics, Basel, Switzerland 2Case Western Reserve University, Department of Neurology, Cleveland, USA 3University of Basel, Biozentrum, Basel, Switzerland Abstract: Mutations of DYSF, the gene encoding dysferlin, cause two types of muscular dystrophies: limb-girdle muscu- lar dystrophy type 2B and Miyoshi myopathy. Recent work suggests a role of dysferlin in membrane repair and demon- strates that defective membrane repair is a novel mechanism of muscle degeneration. We used the tandem affinity purifi- cation method for the purification of proteins interacting with dysferlin. Three interacting partners were identified by this method (striatin, adaptin alpha, utrophin) and were confirmed by co-immunoprecipitations. All three proteins play a role in vesicle trafficking. Knowing the interacting partners of dysferlin will help to understand how muscle cells repair tears in the sarcolemma and will give a deeper insight into this very important cell function. At the same time the identified proteins could serve as potential candidates for other muscular dystrophies and muscle-related diseases with unknown ae- tiology. INTRODUCTION this machinery. These proteins could serve as potential can- didate for other muscular dystrophies and muscle-diseases Dysferlin is a member of a mammalian gene family shar- with unknown aetiology. Therefore, we attempted to identify ing homology with the Caenorhabditis elegans spermato- the interacting partners of dysferlin and used the tandem genesis factor fer-1 gene, which mediates vesicle fusion to affinity purification (TAP) method for this purpose. -

Mapping Transmembrane Binding Partners for E-Cadherin Ectodomains
SUPPLEMENTARY INFORMATION TITLE: Mapping transmembrane binding partners for E-cadherin ectodomains. AUTHORS: Omer Shafraz 1, Bin Xie 2, Soichiro Yamada 1, Sanjeevi Sivasankar 1, 2, * AFFILIATION: 1 Department of Biomedical Engineering, 2 Biophysics Graduate Group, University of California, Davis, CA 95616. *CORRESPONDING AUTHOR: Sanjeevi Sivasankar, Tel: (530)-754-0840, Email: [email protected] Figure S1: Western blots a. EC-BioID, WT and Ecad-KO cell lysates stained for Ecad and tubulin. b. HRP-streptavidin staining of biotinylated proteins eluted from streptavidin coated magnetic beads incubated with cell lysates of EC-BioID with (+) and without (-) exogenous biotin. c. C-BioID, WT and Ecad-KO cell lysates stained for Ecad and tubulin. d. HRP-streptavidin staining of biotinylated proteins eluted from streptavidin coated magnetic beads incubated with cell lysates of C-BioID with (+) and without (-) exogenous biotin. (+) Biotin (-) Biotin Sample 1 Sample 2 Sample 3 Sample 4 Sample 1 Sample 2 Sample 3 Sample 4 Percent Percent Percent Percent Percent Percent Percent Percent Gene ID Coverage Coverage Coverage Coverage Coverage Coverage Coverage Coverage CDH1 29.6 31.4 41.1 36.5 10.8 6.7 28.8 29.1 DSG2 26 14.6 45 37 0.8 1.9 1.6 18.7 CXADR 30.2 26.2 32.7 27.1 0.0 0.0 0.0 6.9 EFNB1 24.3 30.6 24 30.3 0.0 0.0 0.0 0.0 ITGA2 16.5 22.2 30.1 33.4 1.1 1.1 5.2 7.2 CDH3 21.8 9.7 20.6 25.3 1.3 1.3 0.0 0.0 ITGB1 11.8 16.7 23.9 20.3 0.0 2.9 8.5 5.8 DSC3 9.7 7.5 11.5 13.3 0.0 0.0 2.6 0.0 EPHA2 23.2 31.6 31.6 30.5 0.8 0.0 0.0 5.7 ITGB4 21.8 27.8 33.1 30.7 0.0 1.2 3.9 4.4 ITGB3 23.5 22.2 26.8 24.7 0.0 0.0 5.2 9.1 CDH6 22.8 18.1 28.6 24.3 0.0 0.0 0.0 9.1 CDH17 8.8 12.4 20.7 18.4 0.0 0.0 0.0 0.0 ITGB6 12.7 10.4 14 17.1 0.0 0.0 0.0 1.7 EPHB4 11.4 8.1 14.2 16.3 0.0 0.0 0.0 0.0 ITGB8 5 10 15 17.6 0.0 0.0 0.0 0.0 ITGB5 6.2 9.5 15.2 13.8 0.0 0.0 0.0 0.0 EPHB2 8.5 4.8 9.8 12.1 0.0 0.0 0.0 0.0 CDH24 5.9 7.2 8.3 9 0.0 0.0 0.0 0.0 Table S1: EC-BioID transmembrane protein hits. -

Induction of Therapeutic Tissue Tolerance Foxp3 Expression Is
Downloaded from http://www.jimmunol.org/ by guest on October 2, 2021 is online at: average * The Journal of Immunology , 13 of which you can access for free at: 2012; 189:3947-3956; Prepublished online 17 from submission to initial decision 4 weeks from acceptance to publication September 2012; doi: 10.4049/jimmunol.1200449 http://www.jimmunol.org/content/189/8/3947 Foxp3 Expression Is Required for the Induction of Therapeutic Tissue Tolerance Frederico S. Regateiro, Ye Chen, Adrian R. Kendal, Robert Hilbrands, Elizabeth Adams, Stephen P. Cobbold, Jianbo Ma, Kristian G. Andersen, Alexander G. Betz, Mindy Zhang, Shruti Madhiwalla, Bruce Roberts, Herman Waldmann, Kathleen F. Nolan and Duncan Howie J Immunol cites 35 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription http://www.jimmunol.org/content/suppl/2012/09/17/jimmunol.120044 9.DC1 This article http://www.jimmunol.org/content/189/8/3947.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2012 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of October 2, 2021. -

Plasticity and the Impact of Increasing Temperature on a Tropical Ectotherm
Georgia Southern University Digital Commons@Georgia Southern Electronic Theses and Dissertations Graduate Studies, Jack N. Averitt College of Spring 2020 Plasticity and the Impact of Increasing Temperature on a Tropical Ectotherm Adam A. Rosso Follow this and additional works at: https://digitalcommons.georgiasouthern.edu/etd Part of the Bioinformatics Commons, Comparative and Evolutionary Physiology Commons, Evolution Commons, Integrative Biology Commons, Other Ecology and Evolutionary Biology Commons, and the Other Genetics and Genomics Commons Recommended Citation Rosso, Adam A., "Plasticity and the Impact of Increasing Temperature on a Tropical Ectotherm" (2020). Electronic Theses and Dissertations. 2069. https://digitalcommons.georgiasouthern.edu/etd/2069 This thesis (open access) is brought to you for free and open access by the Graduate Studies, Jack N. Averitt College of at Digital Commons@Georgia Southern. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons@Georgia Southern. For more information, please contact [email protected]. PLASTICITY AND THE IMPAPCT OF INCREASING TEMPERATURE ON A TROPICAL ECTOTHERM by ADAM A. ROSSO (Under the direction of Christian L. Cox) ABSTRACT Organisms may respond to climate change through behavior, genetic adaptation, and/or phenotypic plasticity. Tropical ectotherms are thought to be especially vulnerable to climate change because most have a narrow range of thermal tolerance while living close to their upper thermal tolerance limits. Additionally, many tropical species live in closed-canopy forests, which provide homogenous thermal landscapes that prevent behavioral compensation for stressfully warm temperatures. Finally, tropical ectotherms are thought to have decreased capacity for phenotypic plasticity because they have evolved in thermally stable environments. -

(12) Patent Application Publication (10) Pub. No.: US 2017/0067.111 A1 Rothenberg Et Al
US 20170067.111A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0067.111 A1 Rothenberg et al. (43) Pub. Date: Mar. 9, 2017 (54) EVALUATION OF EOSINOPHILIC 2007, now Pat. No. 8,030.003, filed as application ESOPHAGITIS No. PCT/US2005/044456 on Dec. 7, 2005. (71) Applicant: CHILDRENS HOSPITAL (60) Provisional application No. 60/633,909, filed on Dec. MEDICAL CENTER, Cincinnati, OH 7, 2004. (US) Publication Classification (72) Inventors: Marc E. Rothenberg, Cincinnati, OH (51) Int. Cl. (US); Carrie Blanchard, Cincinnati, CI2O I/68 (2006.01) OH (US) A6II 3/56 (2006.01) (52) U.S. Cl. (21) Appl. No.: 15/340,282 CPC ............. CI2O 1/6883 (2013.01); A61 K3I/56 Filed: Nov. 1, 2016 (2013.01); C12O 2600/158 (2013.01); C12O (22) 2600/16 (2013.01) Related U.S. Application Data (57) ABSTRACT (63) Continuation of application No. 12/492.456, filed on Jun. 26, 2009, now abandoned, which is a continua A method to evaluate eosinophilic esophagitis based on tion of application No. 1 1/721,127, filed on Jun. 7, information in an eosinophilic esophagitis transcriptome. EE FO GENE SE GENEBANK SY-B NBER MBER N. EE RESPONDERS CHANGES PKB NMOO6952 C NMO280 SH2OB NMO53282 F NM000204 CA34.54 EM N-OOOO8707 AADAC2 NM2O7365 CH3 NMOO276 SNX 9 NMO 14758 AR NM000045 PNLFRF3 NMOOOO 709 SS - NM00942 Patent Application Publication Mar. 9, 2017. Sheet 1 of 4 US 2017/0067.111A1 18O, OO 60.00 40.00 2O, OO OC, OO 8O, OO 60.00 AO, OO 2O. OO OOO NORMA EE F. -

ERK1/2 MAP Kinases: Structure, Function, and Regulation
Pharmacological Research 66 (2012) 105–143 Contents lists available at SciVerse ScienceDirect Pharmacological Research jo urnal homepage: www.elsevier.com/locate/yphrs Invited review ERK1/2 MAP kinases: Structure, function, and regulation ∗ Robert Roskoski Jr. Blue Ridge Institute for Medical Research, 3754 Brevard Road, Suite 116, Box 19, Horse Shoe, NC 28742, USA a r t i c l e i n f o a b s t r a c t Article history: ERK1 and ERK2 are related protein-serine/threonine kinases that participate in the Ras-Raf-MEK-ERK Received 19 April 2012 signal transduction cascade. This cascade participates in the regulation of a large variety of processes Accepted 20 April 2012 including cell adhesion, cell cycle progression, cell migration, cell survival, differentiation, metabolism, proliferation, and transcription. MEK1/2 catalyze the phosphorylation of human ERK1/2 at Tyr204/187 Keywords: and then Thr202/185. The phosphorylation of both tyrosine and threonine is required for enzyme acti- Docking site vation. Whereas the Raf kinase and MEK families have narrow substrate specificity, ERK1/2 catalyze Nucleoporin the phosphorylation of hundreds of cytoplasmic and nuclear substrates including regulatory molecules Protein kinase inhibitor and transcription factors. ERK1/2 are proline-directed kinases that preferentially catalyze the phos- Protein phosphatase Raf phorylation of substrates containing a Pro-Xxx-Ser/Thr-Pro sequence. Besides this primary structure Ras requirement, many ERK1/2 substrates possess a D-docking site, an F-docking site, or both. A variety of scaffold proteins including KSR1/2, IQGAP1, MP1, -Arrestin1/2 participate in the regulation of the ERK1/2 MAP kinase cascade. -

Reproductionresearch
REPRODUCTIONRESEARCH NUP50 is necessary for the survival of primordial germ cells in mouse embryos Eunsook Park1,2, Bobae Lee1, Bruce E Clurman3 and Keesook Lee1 1School of Biological Sciences and Technology and 2Korea Basic Science Institute Gwangju Center, Chonnam National University, Gwangju 500-757, Republic of Korea and 3Divisions of Clinical Research and Human Biology, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA Correspondence should be addressed to K Lee; Email: [email protected] Abstract Nucleoporin 50 kDa (NUP50), a component of the nuclear pore complex, is highly expressed in male germ cells, but its role in germ cells is largely unknown. In this study, we analyzed the expression and function of NUP50 during the embryonic development of germ cells using NUP50-deficient mice. NUP50 was expressed in germ cells of both sexes at embryonic day 15.5 (E15.5), E13.5, and E12.5. In addition, NUP50 expression was also detected in primordial germ cells (PGCs) migrating into the genital ridges at E9.5. The gonads of Nup50K/K embryos of both sexes contained few PGCs at both E11.5 and E12.5 and no developing germ cells at E15.5. The migratory PGCs in Nup50K/K embryos at E9.5 showed increased apoptosis but a normal rate of proliferation, resulting in the progressive loss of germ cells at later stages. Taken together, these results suggest that NUP50 plays an essential role in the survival of PGCs during embryonic development. Reproduction (2016) 151 51–58 Introduction remaining PGCs in the midline structures during the late stages of development (Runyan et al. -

Detection of Tissue-Specific Genes and Computational Analysis of Testis
Detection of tissue-specific genes and Title computational analysis of testis-specific gene expression regulatory regions Author(s) 山下, 明史 Citation Issue Date Text Version ETD URL http://hdl.handle.net/11094/2599 DOI rights Note Osaka University Knowledge Archive : OUKA https://ir.library.osaka-u.ac.jp/ Osaka University Detection of tissue-specific genes and computational analysis of testis-specific gene expression regulatory regions (組織特異的遺伝子の検出と精巣特異的遺伝子の 発現制御領域のコンピュータを使った解析) by Akifumi Yamashita Department of Genome Informatics, Genome Information Research Center, Research Institute for Microbial Diseases, Osaka University, 3-1 Yamadaoka, Suita, Osaka 565-0871, Japan February, 2009 1/97 Contents LIST of ABBREVIATIONS 5 ABSTRACT 6 1 INTRODUCTION 7 2 METHODS 10 2.1 Detection of tissue-specific genes 10 2.2 Annotation of tissue-specific genes 10 2.3 Extracting the 5′-regulatory regions 11 2.4 Selection of over-represented 8-mers 11 2.5 Comparison of the 8-mer frequency within regulatory regions of 13 testis-specific genes with those of non-testis-specific genes. 3 RESULTS 14 3.1 Detection of tissue-specific genes 14 3.2 Selection of testis-specific genes which can be used for the 14 following analysis 3.3 Classification of the over-represented 8-mers 15 3.4 Conservations of the flanking region of highlighted 8-mers 16 2/97 4 DISCUSSION 18 ACKNOWLEDGEMENTS 23 REFERENCES 24 TABLES 27 Table 1 - Number of tissue-specific genes 27 Table 2 - Classification of 117 representative 8-mers on the basis of their 28 testis association level Table 3 - 8-mers which appeared near by the other 8-mers 31 FIGURES 32 Fig. -

WRAP Theses Panetta 2021.Pdf
A Thesis Submitted for the Degree of PhD at the University of Warwick Permanent WRAP URL: http://wrap.warwick.ac.uk/154378 Copyright and reuse: This thesis is made available online and is protected by original copyright. Please scroll down to view the document itself. Please refer to the repository record for this item for information to help you to cite it. Our policy information is available from the repository home page. For more information, please contact the WRAP Team at: [email protected] warwick.ac.uk/lib-publications Characterization of O-GlcNAc signalling in human placenta by Pamela Panetta Warwick Medical School University of Warwick A thesis submitted for the degree of Doctor of Philosophy January, 2021 TABLE OF CONTENTS List of figures 7 List of tables 10 Acknowledgments 11 Declaration 12 Abstract 13 List of abbreviations 14 Chapter 1 - Introduction 18 1.1 Human placenta 19 1.1.1 Formation of human placenta 19 1.1.2 Signalling pathways regulating syncytiotrophoblast formation 21 1.1.2.1 Experimental models to study human trophoblast differentiation 24 1.1.3 Functions of human placenta: role of placental barrier 27 1.1.3.1 Transport of macronutrients glucose, amino acids and lipids 29 1.1.3.2 Protective function 31 1.1.3.3 Endocrine function 31 1.1.4 Placental mechanisms of in utero programming: contribution of maternal overnutrition and stress 35 1.1.4.1 Epidemiologic evidence of in utero programming 35 1.1.4.2 Role of placental nutrient transport in fetal programming 37 1.1.4.3 Placental nutrient-sensing system in fetal -
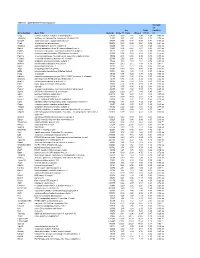
Table S-1 Mpkccd Transcriptome
Table S1. mpkCCD Cell Transcriptomes. Correlati on Ori. Ratio Coefficie Gene Symbol Gene Title GeneID Clone 11 Clone Clone 2 (11/2) nt Copg coatomer protein complex, subunit gamma 54161 1.99 1.81 2.48 0.80 -0.49 ns Atp6v0d1 ATPase, H+ transporting, lysosomal V0 subunit D1 11972 3.37 2.87 3.00 1.12 0.74 ns Golga7 golgi autoantigen, golgin subfamily a, 7 57437 3.03 3.40 3.58 0.84 -0.34 ns Psph phosphoserine phosphatase 100678 0.38 0.66 0.64 0.59 -0.61 ns Trappc4 trafficking protein particle complex 4 60409 1.17 1.12 1.08 1.08 0.64 ns Dpm2 dolichol-phosphate (beta-D) mannosyltransferase 2 13481 0.74 0.87 0.81 0.91 -0.31 ns Psmb5 proteasome (prosome, macropain) subunit, beta type 5 19173 2.92 3.00 2.92 0.99 0.32 ns Dhrs1 dehydrogenase/reductase (SDR family) member 1 52585 0.74 0.68 0.88 0.83 -0.18 ns Ppm1a protein phosphatase 1A, magnesium dependent, alpha isoform 19042 2.43 2.60 2.96 0.82 -0.53 ns Psenen presenilin enhancer 2 homolog (C. elegans) 66340 2.52 2.88 3.80 0.66 -0.77 ns Anapc1 anaphase promoting complex subunit 1 17222 1.16 1.33 1.61 0.72 -0.33 ns Mrpl43 mitochondrial ribosomal protein L43 94067 2.15 2.15 1.84 1.16 0.91 * Nmt1 N-myristoyltransferase 1 18107 3.21 2.71 3.36 0.95 0.31 ns Atg5 autophagy-related 5 (yeast) 11793 0.70 0.73 0.66 1.05 -0.23 ns Mtif2 mitochondrial translational initiation factor 2 76784 1.16 1.29 1.19 0.97 -0.36 ns Psap prosaposin 19156 8.07 7.25 8.37 0.96 0.02 ns Ube2g1 ubiquitin-conjugating enzyme E2G 1 (UBC7 homolog, C. -

Types of Nuclear Localization Signals and Mechanisms of Protein Import
Lu et al. Cell Commun Signal (2021) 19:60 https://doi.org/10.1186/s12964-021-00741-y REVIEW Open Access Types of nuclear localization signals and mechanisms of protein import into the nucleus Juane Lu1, Tao Wu1, Biao Zhang1, Suke Liu1, Wenjun Song1, Jianjun Qiao2,3,4* and Haihua Ruan1* Abstract Nuclear localization signals (NLS) are generally short peptides that act as a signal fragment that mediates the trans- port of proteins from the cytoplasm into the nucleus. This NLS-dependent protein recognition, a process necessary for cargo proteins to pass the nuclear envelope through the nuclear pore complex, is facilitated by members of the importin superfamily. Here, we summarized the types of NLS, focused on the recently reported related proteins containing nuclear localization signals, and briefy summarized some mechanisms that do not depend on nuclear localization signals into the nucleus. Keywords: Nuclear localization signal, Nuclear pore complex, Importin Background a permeability barrier between the cytoplasm and nucle- One of the characteristic features of eukaryotic cells oplasm [5]. Te main structural components of the NPC are membrane-bound functional organelles such as the include the central channel, the cytoplasmic ring moiety nucleus, mitochondria, golgi apparatus, and others, and cytoplasmic flaments, and the nuclear ring moiety which are surrounded by cytoplasm. For cells to function and nuclear basket [6]. Te NPC has eightfold rotational normally, organelle proteins synthesized in the cytoplasm symmetry. Each NPC is connected to the inner and outer must be selectively and efciently transported into their nuclear membranes by symmetrical 8 molecular spoke destination compartments where they can exert their proteins, and the 8 molecular spoke proteins surround physiological functions [1].