19770005666.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Material Safety Data Sheet Prepared to US OSHA, CMA, ANSI, Canadian WHMIS and EU Standards
MSDS NUMBER: 1201 Fluorine Excimer Laser Mix (1.0% or less Fluorine) Material Safety Data Sheet Prepared to US OSHA, CMA, ANSI, Canadian WHMIS and EU Standards SECTION 1. PRODUCT IDENTIFICATION PRODUCT NAME: Fluorine Excimer Laser Mix (1.0% or less Fluorine) US DOT NAME: Compressed Gas, n.o.s. (Fluorine, other gas) UN 1956 (see Sec. 14) CHEMICAL NAME: Mixture of Fluorine (1.0% or Less) and Argon, Krypton or Xenon balance Helium, Neon or Nitrogen FORMULA: Argon = Ar; Fluorine = F2; Helium = He; Krypton = Kr; Neon = Ne; Nitrogen = N2; SYNONYMS: Not Applicable US MANUFACTURER: Linde LLC 575 Mountain Ave. Murray Hill, NJ 07974 Phone: 908-464-8100 lindeus.com 24 HOUR EMERGENCY CONTACT, CHEMTREC: 800/424-9300, 703/527-3887 For additional product information contact your customer service representative. PRODUCT USE: In Excimer Laser and Research and Development ALL WHMIS required information is included in appropriate sections based on the ANSI Z400.1-2004 format. This product has been classified in accordance with the hazard criteria of the CPR and the MSDS contains all the information required by the CPR. The product is also classified per all applicable EU Directives through EC 1907: 2006 SECTION 2. HAZARD IDENTIFICATION EU LABELING AND CLASSIFICATION: This gas mixture meets the definition of hazardous according to the criteria of the European Union Council Directive 67/548/EEC and based on current guidelines under EC 1907: 2006. EU CLASSIFICATION: Xn (Harmful) EU RISK PHRASES: R: 20; R: 21; R: 36/37/38 EU SAFETY PHRASES: S: (1/2)*; S: 7/9, S: 26, S: 36/37/39, S: 45 See Section 15 for full definition of Risk and Safety Phrases. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

Attached Are the MSDS Lists for Various Chemicals Used During the Wet Plate Collodion / Platinum Palladium Courses at Oxbow
Attached are the MSDS lists for various chemicals used during the Wet Plate Collodion / Platinum Palladium courses at OxBow. Many of the materials for this course are highly toxic and therefore thorough communication and understanding about this aspect of wet plate collodion. Safety is a necessity. It is our first priority and concern to having a productive community throughout our courses. Please contact us should you have any questions or concerns, we will be happy to answer. Thank you, Jaclyn Silverman and Robert Clarke-Davis MSDS Lists: Wet-plate collodion * you will be required to use nitrile gloves, there is no exception to this rule. SAFETY DATA SHEET Preparation Date: No data available Revision Date: 6/17/2015 Revision Number: G1 Product identifier Product code: CO120 Product Name: COLLODION, USP Other means of identification Synonyms: No information available CAS #: Mixture RTECS # Not available CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: No information available. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000 Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Acute toxicity - Oral Category 4 Skin corrosion/irritation Category 2 Serious eye damage/eye irritation Category 2 Reproductive toxicity Category 1B Specific target organ toxicity (single exposure) Category 3 Specific target organ toxicity (repeated exposure) Category 1 Flammable liquids Category 1 Label elements Product code: CO120 Product name: COLLODION, USP 1 / 16 Danger Hazard statements Harmful if swallowed Causes skin irritation Causes serious eye irritation May damage fertility or the unborn child May cause respiratory irritation. -

List of Reactive Chemicals
LIST OF REACTIVE CHEMICALS Chemical Prefix Chemical Name Reactive Reactive Reactive CAS# Chemical Chemical Chemical Stimulus 1 Stimulus 2 Stimulus 3 111-90-0 "CARBITOL" SOLVENT D 111-15-9 "CELLOSOLVE" ACETATE D 110-80-5 "CELLOSOLVE" SOLVENT D 2- (2,4,6-TRINITROPHENYL)ETHYL ACETATE (1% IN ACETONE & BENZENE S 12427-38-2 AAMANGAN W 88-85-7 AATOX S 40487-42-1 AC 92553 S 105-57-7 ACETAL D 75-07-0 ACETALDEHYDE D 105-57-7 ACETALDEHYDE, DIETHYL ACETAL D 108-05-4 ACETIC ACID ETHENYL ESTER D 108-05-4 ACETIC ACID VINYL ESTER D 75-07-0 ACETIC ALDEHYDE D 101-25-7 ACETO DNPT T 126-84-1 ACETONE DIETHYL ACETAL D 108-05-4 ACETOXYETHYLENE D 108-05-4 1- ACETOXYETHYLENE D 37187-22-7 ACETYL ACETONE PEROXIDE, <=32% AS A PASTE T 37187-22-7 ACETYL ACETONE PEROXIDE, <=42% T 37187-22-7 ACETYL ACETONE PEROXIDE, >42% T S 644-31-5 ACETYL BENZOYL PEROXIDE (SOLID OR MORE THAN 45% IN SOLUTION) T S 644-31-5 ACETYL BENZOYL PEROXIDE, <=45% T 506-96-7 ACETYL BROMIDE W 75-36-5 ACETYL CHLORIDE W ACETYL CYCLOHEXANE SULFONYL PEROXIDE (>82% WITH <12% WATER) T S 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=32% T 3179-56-4 ACETYL CYCLOHEXANE SULFONYL PEROXIDE, <=82% T 674-82-8 ACETYL KETENE (POISON INHALATION HAZARD) D 110-22-5 ACETYL PEROXIDE, <=27% T 110-22-5 ACETYL PEROXIDE, SOLID, OR MORE THAN 27% IN SOLUTION T S 927-86-6 ACETYLCHOLINE PERCHLORATE O S 74-86-2 ACETYLENE D 74-86-2 ACETYLENE (LIQUID) D ACETYLENE SILVER NITRATE D 107-02-08 ACRALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACROLEIC ACID D 107-02-08 ACROLEIN, INHIBITED (POISON INHALATION HAZARD) D 107-02-08 ACRYLALDEHYDE (POISON INHALATION HAZARD) D 79-10-7 ACRYLIC ACID D 141-32-2 ACRYLIC ACID BUTYL ESTER D 140-88-5 ACRYLIC ACID ETHYL ESTER D 96-33-3 ACRYLIC ACID METHYL ESTER D Stimulus - Stimuli is the thermal, physical or chemical input needed to induce a hazardous reaction. -

Inorganic Chemistry
INORGANIC CHEMISTRY Saito Inorganic Chemistry Saito This text is disseminated via the Open Education Resource (OER) LibreTexts Project (https://LibreTexts.org) and like the hundreds of other texts available within this powerful platform, it freely available for reading, printing and "consuming." Most, but not all, pages in the library have licenses that may allow individuals to make changes, save, and print this book. Carefully consult the applicable license(s) before pursuing such effects. Instructors can adopt existing LibreTexts texts or Remix them to quickly build course-specific resources to meet the needs of their students. Unlike traditional textbooks, LibreTexts’ web based origins allow powerful integration of advanced features and new technologies to support learning. The LibreTexts mission is to unite students, faculty and scholars in a cooperative effort to develop an easy-to-use online platform for the construction, customization, and dissemination of OER content to reduce the burdens of unreasonable textbook costs to our students and society. The LibreTexts project is a multi-institutional collaborative venture to develop the next generation of open-access texts to improve postsecondary education at all levels of higher learning by developing an Open Access Resource environment. The project currently consists of 13 independently operating and interconnected libraries that are constantly being optimized by students, faculty, and outside experts to supplant conventional paper-based books. These free textbook alternatives are organized within a central environment that is both vertically (from advance to basic level) and horizontally (across different fields) integrated. The LibreTexts libraries are Powered by MindTouch® and are supported by the Department of Education Open Textbook Pilot Project, the UC Davis Office of the Provost, the UC Davis Library, the California State University Affordable Learning Solutions Program, and Merlot. -
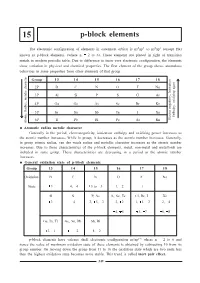
P-Block Elements
15 p-block elements The electronic configuration of elements in outermost orbital is ns2np1 to ns2np6 (except He) known as p-block elements. (where n = 2 to 6). These elements are placed in right of transition metals in modern periodic table. Due to difference in inner core electronic configuration, the elements show variation in physical and chemical properties. The first element of the group shows anomalous behaviour in some properties from other elements of that group. Group 13 14 15 16 17 18 2P BC NO F Ne 3P Al Si P S Cl Ar , Ionisation 4P Ga Ge As Se Br Kr , oxidizing agent 5P In Sn Sb Te I Xe Enthalpy 6P Tl Pb Bi Po At Rn Electro negativity Atomic Radius, metalic character Atomatic radius metalic character Generally in the period, electronegativity, ionization enthalpy and oxidising power increases as the atomic number increases. While in group, it decreases as the atomic number increases. Generally, in group atomic radius, van der waals radius and metallic character increases as the atomic number increases. Due to these characteristics of the p-block elements, metal, non-metal and metalloids are included in same group. These characteristics are decreasing in a period as the atomic number increases. General oxidation state of p-block elements Group 13 14 15 16 17 18 Oxidation BC NO F Ne State +3 +4, -4 +5 to -3 -1, -2 -1 - Al Si P, As S, Se, Te Cl, Br, I Xe +3 +4 +3, +5, -3 -2, +2 -1, +1, +3 +2, +4 +4, +6 +5, +7 +6, +8 Ga, In, Tl Ge, Sn, Pb Sb, Bi +3, +1 +4, +2 +4, +2 p-block elements have valence shell electronic configuration ns2np16 where n = 2 to 6 and hence the value of maximum oxidation state of these elements is obtained by subtracting 10 from its group number. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Witch Hazel Extract, Double Distilled
SAFETY DATA SHEET Preparation Date: 11/11/2015 Revision Date: 11/11/2015 Revision Number: G1 1. IDENTIFICATION Product identifier Product code: WI105 Product Name: WITCH HAZEL EXTRACT, DOUBLE DISTILLED Other means of identification Synonyms: Amamelide, extract Hamamelis virginiana, extract CAS #: Mixture RTECS # MG8328500 CI#: Not available Recommended use of the chemical and restrictions on use Recommended use: No information available. Uses advised against No information available Supplier: Spectrum Chemical Mfg. Corp 14422 South San Pedro St. Gardena, CA 90248 (310) 516-8000 Order Online At: https://www.spectrumchemical.com Emergency telephone number Chemtrec 1-800-424-9300 Contact Person: Martin LaBenz (West Coast) Contact Person: Ibad Tirmiz (East Coast) 2. HAZARDS IDENTIFICATION Classification This chemical is considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Serious eye damage/eye irritation Category 2B Flammable liquids Category 3 Label elements Product code: WI105 Product name: WITCH HAZEL 1 / 14 EXTRACT, DOUBLE DISTILLED Warning Hazard statements Causes eye irritation Flammable liquid and vapor Hazards not otherwise classified (HNOC) Not Applicable Other hazards Can burn with an invisible flame Precautionary Statements - Prevention Wash face, hands and any exposed skin thoroughly after handling Keep away from heat/sparks/open flames/hot surfaces. — No smoking Keep container tightly closed Ground/bond container and receiving equipment Use explosion-proof electrical/ventilating/lighting/ .? /equipment Use only non-sparking tools Take precautionary measures against static discharge Wear protective gloves Wear eye/face protection In case of fire: Use CO2, dry chemical, or foam to extinguish. IF IN EYES: Rinse cautiously with water for several minutes. -

2 1 0 Material Safety Data Sheet
He a lt h 1 2 Fire 2 1 0 Re a c t iv it y 0 Pe rs o n a l Pro t e c t io n H Material Safety Data Sheet Methylene Blue Solution, Loeffler MSDS Section 1: Chemical Product and Company Identification Product Name: Methylene Blue Solution, Loeffler Contact Information: Catalog Codes: SLM3328 Sciencelab.com, Inc. 14025 Smith Rd. CAS#: Mixture. Houston, Texas 77396 RTECS: Not applicable. US Sales: 1-800-901-7247 International Sales: 1-281-441-4400 TSCA: TSCA 8(b) inventory: Potassium hydroxide; Ethyl alcohol 200 Proof; Water Order Online: ScienceLab.com CI#: Not applicable. CHEMTREC (24HR Emergency Telephone), call: 1-800-424-9300 Synonym: Methylene Blue-Loeffler's Alkaline International CHEMTREC, call: 1-703-527-3887 Chemical Name: Not applicable. For non-emergency assistance, call: 1-281-441-4400 Chemical Formula: Not applicable. Section 2: Composition and Information on Ingredients Composition: Name CAS # % by Weight Potassium hydroxide 1310-58-3 <1 Ethyl alcohol 200 Proof 64-17-5 25-30 Water 7732-18-5 70-75 Methylene Blue CI52015 7220-79-3 <1 Toxicological Data on Ingredients: Ethyl alcohol 200 Proof: ORAL (LD50): Acute: 7060 mg/kg [Rat.]. 3450 mg/kg [Mouse]. VAPOR (LC50): Acute: 20000 ppm 8 hours [Rat]. 39000 mg/m 4 hours [Mouse]. Section 3: Hazards Identification Potential Acute Health Effects: Hazardous in case of skin contact (irritant, permeator), of eye contact (irritant), of ingestion. Potential Chronic Health Effects: CARCINOGENIC EFFECTS: Classified PROVEN by State of California Proposition 65 [Ethyl alcohol 200 Proof]. Classified A4 (Not classifiable for human or animal.) by ACGIH [Ethyl alcohol 200 Proof]. -

=« Y * MASS SPECTROMETRIC STUDIES of the SYNTHESIS
"In presenting the dissertation as a partial fulfillment of the requirements for an advanced degree from the Georgia Institute of Technology, I agree that the Library of the Institution shall make it available for inspection and circulation in accordance with its regulations governing materials of this type. I agree that permission to copy from, or to publish from, this dissertation may be granted by the professor under whose direction it was written, or, in his absence, by the dean of the Graduate Division when such copying or publication is solely for scholarly purposes and does not involve potential financial gain. It is understood that any copying from, or publication of, this dissertation which involves potential financial gain will not be allowed without written permission, "-- -. • i -=« y * MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES A THESIS Presented to The Faculty of the Graduate Division by Thomas Joseph Malone In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the School of Chemical Engineering Georgia Institute of Technology February, 1966 MASS SPECTROMETRIC STUDIES OF THE SYNTHESIS, REACTIVITY, AND ENERGETICS OF THE OXYGEN FLUORIDES AT CRYOGENIC TEMPERATURES Approved; / • ' "/v Date approved by Chairma n:\PuJl 3 lUt, 11 ACKNOWLEDGMENTS I am deeply indebted to Dr0 Ho Ao McGee, Jr0 for his encouragement, advice^ and assistance throughout both my graduate and undergraduate studieso Many hours of discussion and willing -

Nitrogen Oxychlorides : a Bibliography on Data for Physical and Chemical
NBS Publl - cations NArL INST. OF STAND & TECH NBS SPECIAL PUBLICATION 478 U.S. DEPARTMENT OF COMMERCE / National Bureau of Standards NITROGEN OXYCHLORIDES: A Bibliography on Data for Physical and Chemical Properties of CUNO, CLNO2, and CdNOs NATIONAL BUREAU OF STANDARDS The National Bureau of Standards^ was established by an act of Congress March 3, 1901. The Bureau's overall goal is to' strengthen and advance the Nation's science and technology and facilitate their effective application for public benefit. To this end, the Bureau conducts research and provides: (1) a basis for the Nation's physical measurement system, (2) scientific and technological services for industry and government, (3) a technical basis for equity in trade, and (4) technical services to pro- mote public safety. The Bureau consists of the Institute for Basic Standards, the Institute for Materials Research, the Institute! for Applied Technology, the Institute for Computer Sciences and Technology, the Office for Information Programs, and the Office of Experimental Technology Incentives Program. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consist- ent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essen-' tial services leading to accurate and uniform physical measurements throughout the Nation's scientific community, industry,! and commerce. The Institute consists of the Office of Measurement Services, and the following center and divisions: -

Pp-03-25-New Dots.Qxd 10/23/02 2:41 PM Page 611
pp-03-25-new dots.qxd 10/23/02 2:41 PM Page 611 NICKEL CARBONATE 611 NICKEL CARBONATE [3333-67-3] Formula: NiCO3; MW 118.72 Two basic carbonates are known. They are 2NiCO3•3Ni(OH)2•4H2O [29863- 10-3], and NiCO3•2Ni(OH)2 [12607-70-4], MW 304.17. The second form occurs in nature as a tetrahydrate, mineral, zaratite. Commercial nickel car- bonate is usually the basic salt, 2NiCO3•3Ni(OH)2•4H2O. Uses Nickel carbonate is used to prepare nickel catalysts and several specialty compounds of nickel. It also is used as a neutralizing agent in nickel plating solutions. Other applications are in coloring glass and in the manufacture of ceramic pigments. Physical Properties NiCO3: Light green rhombohedral crystals; decomposes on heating; practi- cally insoluble in water, 93 mg/L at 25°C; dissolves in acids. 2NiCO3•3Ni(OH)2•4H2O: Light green crystals or brown powder; decom- poses on heating; insoluble in water; decomposes in hot water; soluble in acids and in ammonium salts solutions. Zaratite: Emerald greed cubic crystals; density 2.6 g/cm3; insoluble in water; soluble in ammonia and dilute acids. Thermochemical Properties ∆Ηƒ° (NiCO3) –140.6 kcal/mol Preparation Anhydrous nickel carbonate is produced as a precipitate when calcium car- bonate is heated with a solution of nickel chloride in a sealed tube at 150°C. Alternatively, treating nickel powder with ammonia and carbon dioxide fol- lowed by boiling off ammonia yields pure carbonate. When sodium carbonate is added to a solution of Ni(II) salts, basic nickel carbonate precipitates out in impure form.