SLC Transporters As Therapeutic Targets: Emerging Opportunities
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Small-Molecule Inhibitors of Pendrin Potentiate the Diuretic Action of Furosemide
BASIC RESEARCH www.jasn.org Small-Molecule Inhibitors of Pendrin Potentiate the Diuretic Action of Furosemide Onur Cil, Peter M. Haggie, Puay-wah Phuan, Joseph-Anthony Tan, and Alan S. Verkman Departments of Medicine and Physiology, University of California San Francisco, San Francisco, California ABSTRACT 2 2 Pendrin is a Cl /HCO3 exchanger expressed in type B and non-A, non-B intercalated cells in the distal 2 nephron, where it facilitates Cl absorption and is involved in Na+ absorption and acid-base balance. Pendrin-knockout mice show no fluid-electrolyte abnormalities under baseline conditions, although mice 2 with double knockout of pendrin and the Na+/Cl cotransporter (NCC) manifest profound salt wasting. Thus, pendrin may attenuate diuretic-induced salt loss, but this function remains unconfirmed. To clarify the physiologic role of pendrin under conditions not confounded by gene knockout, and to test the potential utility of pendrin inhibitors for diuretic therapy, we tested in mice a small-molecule pendrin inhibitor identified from a high-throughput screen. In vitro, a pyrazole-thiophenesulfonamide, PDSinh- 2 C01, inhibited Cl /anion exchange mediated by mouse pendrin with a 50% inhibitory concentration of 1–3 mM, without affecting other major kidney tubule transporters. Administration of PDSinh-C01 to mice at predicted therapeutic doses, determined from serum and urine pharmacokinetics, did not affect urine output, osmolality, salt excretion, or acid-base balance. However, in mice treated acutely with furosemide, + 2 administration of PDSinh-C01 produced a 30% increase in urine output, with increased Na and Cl ex- cretion. In mice treated long term with furosemide, in which renal pendrin is upregulated, PDSinh-C01 produced a 60% increase in urine output. -

Iron Transport Proteins: Gateways of Cellular and Systemic Iron Homeostasis
Iron transport proteins: Gateways of cellular and systemic iron homeostasis Mitchell D. Knutson, PhD University of Florida Essential Vocabulary Fe Heme Membrane Transport DMT1 FLVCR Ferroportin HRG1 Mitoferrin Nramp1 ZIP14 Serum Transport Transferrin Transferrin receptor 1 Cytosolic Transport PCBP1, PCBP2 Timeline of identification in mammalian iron transport Year Protein Original Publications 1947 Transferrin Laurell and Ingelman, Acta Chem Scand 1959 Transferrin receptor 1 Jandl et al., J Clin Invest 1997 DMT1 Gunshin et al., Nature; Fleming et al. Nature Genet. 1999 Nramp1 Barton et al., J Leukocyt Biol 2000 Ferroportin Donovan et al., Nature; McKie et al., Cell; Abboud et al. J. Biol Chem 2004 FLVCR Quigley et al., Cell 2006 Mitoferrin Shaw et al., Nature 2006 ZIP14 Liuzzi et al., Proc Natl Acad Sci USA 2008 PCBP1, PCBP2 Shi et al., Science 2013 HRG1 White et al., Cell Metab DMT1 (SLC11A2) • Divalent metal-ion transporter-1 • Former names: Nramp2, DCT1 Fleming et al. Nat Genet, 1997; Gunshin et al., Nature 1997 • Mediates uptake of Fe2+, Mn2+, Cd2+ • H+ coupled transporter (cotransporter, symporter) • Main roles: • intestinal iron absorption Illing et al. JBC, 2012 • iron assimilation by erythroid cells DMT1 (SLC11A2) Yanatori et al. BMC Cell Biology 2010 • 4 different isoforms: 557 – 590 a.a. (hDMT1) Hubert & Hentze, PNAS, 2002 • Function similarly in iron transport • Differ in tissue/subcellular distribution and regulation • Regulated by iron: transcriptionally (via HIF2α) post-transcriptionally (via IRE) IRE = Iron-Responsive Element Enterocyte Lumen DMT1 Fe2+ Fe2+ Portal blood Enterocyte Lumen DMT1 Fe2+ Fe2+ Fe2+ Fe2+ Ferroportin Portal blood Ferroportin (SLC40A1) • Only known mammalian iron exporter Donovan et al., Nature 2000; McKie et al., Cell 2000; Abboud et al. -

Identification of a Novel Nucleobase-Ascorbate Transporter
bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Identification of a novel Nucleobase-Ascorbate 2 Transporter family member in fish and amphibians 3 Diogo Oliveira*,1, André M. Machado*,1, Tiago Cardoso1, Mónica Lopes-Marques1, L. Filipe 4 C. Castro1,2● and Raquel Ruivo1● 5 1CIIMAR – Interdisciplinary Centre of Marine and Environmental Research, U. Porto – University 6 of Porto, Porto, Portugal 7 2Department of Biology, Faculty of Sciences, U. Porto - University of Porto, Portugal 8 *Equal contribution 9 ●Corresponding authors at: CIIMAR, Terminal de Cruzeiros do Porto de Leixões, Av. General 10 Norton de Matos s/n, 4450-208 Matosinhos, Portugal. Tel.: +351 223 401 831 11 12 Running title: Novel uric acid transporter in fish and amphibians 13 14 15 bioRxiv preprint doi: https://doi.org/10.1101/287870; this version posted December 7, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 16 Abstract:Nucleobase-Ascorbate Transporter (NAT) family includes ascorbic acid, nucleobases 17 and uric acid transporters: with a broad evolutionary distribution. In vertebrates, four members 18 have been previously recognized, the ascorbate transporters Slc23a1 and Slc3a2, the nucleobase 19 transporter Slc23a4 and an orphan transporter Slc23a3. -

Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training
nutrients Review Regulation of Skeletal Muscle Glucose Transport and Glucose Metabolism by Exercise Training Parker L. Evans 1,2,3, Shawna L. McMillin 1,2,3 , Luke A. Weyrauch 1,2,3 and Carol A. Witczak 1,2,3,4,* 1 Department of Kinesiology, East Carolina University, Greenville, NC 27858, USA; [email protected] (P.L.E.); [email protected] (S.L.M.); [email protected] (L.A.W.) 2 Department of Physiology, Brody School of Medicine, East Carolina University, Greenville, NC 27834, USA 3 East Carolina Diabetes & Obesity Institute, East Carolina University, Greenville, NC 27834, USA 4 Department of Biochemistry & Molecular Biology, Brody School of Medicine, East Carolina University, Greenville, NC 27834, USA * Correspondence: [email protected]; Tel.: +1-252-744-1224 Received: 8 September 2019; Accepted: 8 October 2019; Published: 12 October 2019 Abstract: Aerobic exercise training and resistance exercise training are both well-known for their ability to improve human health; especially in individuals with type 2 diabetes. However, there are critical differences between these two main forms of exercise training and the adaptations that they induce in the body that may account for their beneficial effects. This article reviews the literature and highlights key gaps in our current understanding of the effects of aerobic and resistance exercise training on the regulation of systemic glucose homeostasis, skeletal muscle glucose transport and skeletal muscle glucose metabolism. Keywords: aerobic exercise; blood glucose; functional overload; GLUT; hexokinase; insulin resistance; resistance exercise; SGLT; type 2 diabetes; weightlifting 1. Introduction Exercise training is defined as planned bouts of physical activity which repeatedly occur over a duration of time lasting from weeks to years. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
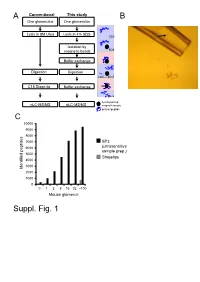
Suppl. Fig. 1 A
A Conventional This study B One glomerulus One glomerulus Lysis in 8M Urea Lysis in 4% SDS SDS Isolation by magnetic beads SDS Buffer exchange Digestion Digestion Try LysC pH>7 C18 Stage tip Buffer exchange pH<4 functionalized nLC-MS/MS nLC-MS/MS magnetic beads protein/peptide C 10000 9000 8000 7000 SP3 6000 (ultrasensitive sample prep.) 5000 Stagetips 4000 3000 Identified peptides 2000 1000 0 0 1 2 8 16 32 ~100 Mouse glomeruli Suppl. Fig. 1 S1. Sample preparation methods. A. Scheme of conventional (C18 Stage Tip) and ultrasensitive proteomic sample preparation methods. B. Manual microdissection and isolation of single glomeruli. The arrow indicates a single isolated glomerulus subjected to proteome analysis. The tip capacity is 10µl. C. Stagetips, a conventional proteomic sample preparation method, is compared with ultrasensitive (SP3) proteome analysis. The indicated amounts of mouse glomeruli were prepared by the respective sample preparation protocols, and peptide numbers after nLC-MS/MS are plotted (all FDR<0.01). A 100 1 Tubule B 80 60 40 Intensity CCD 20 40 0 100 20 S1 TAL 80 0 Tubule proximal tubule Loading... 60 0 Intensity 40 S2 20 -20 control Component 2 (22.5%) S3 0 0 10 20 30 40 50 60 -40 -20 0 20 40 Time (min) Component 2 (47.1%) C D 6000 Mouse proximal tubule 5000 8 4000 3000 7 (iBAQ) Loading... 10 2000 log 6 Number of peptides 1000 0 5 Control mouse mouse mouse human proximal mTAL CCD proximal 0 1500 tubule tubule Rank protein (S1) (S1) R=0.7 E F 8 ACTB Human proximal tubule 8 7 SLC9A3R1 ATP1A1 SLC3A2 SLC25A5 ATP1B1 7 SLC5A2 SLC25A3 CUBN CLTC SLC27A2 SLC5A12 LRP2 6 SLC25A13 SLC25A10 SLC5A1 SLC22A6 6 SLC25A4 Loading.. -

Cell Development and Peripheral Survival Heme Exporter FLVCR Is
Heme Exporter FLVCR Is Required for T Cell Development and Peripheral Survival Mary Philip, Scott A. Funkhouser, Edison Y. Chiu, Susan R. Phelps, Jeffrey J. Delrow, James Cox, Pamela J. Fink and This information is current as Janis L. Abkowitz of September 28, 2021. J Immunol 2015; 194:1677-1685; Prepublished online 12 January 2015; doi: 10.4049/jimmunol.1402172 http://www.jimmunol.org/content/194/4/1677 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/01/09/jimmunol.140217 Material 2.DCSupplemental http://www.jimmunol.org/ References This article cites 44 articles, 11 of which you can access for free at: http://www.jimmunol.org/content/194/4/1677.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 28, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Heme Exporter FLVCR Is Required for T Cell Development and Peripheral Survival Mary Philip,*,† Scott A. -

The Concise Guide to Pharmacology 2019/20
Edinburgh Research Explorer THE CONCISE GUIDE TO PHARMACOLOGY 2019/20 Citation for published version: Cgtp Collaborators 2019, 'THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters', British Journal of Pharmacology, vol. 176 Suppl 1, pp. S397-S493. https://doi.org/10.1111/bph.14753 Digital Object Identifier (DOI): 10.1111/bph.14753 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: British Journal of Pharmacology General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Transporters. British Journal of Pharmacology (2019) 176, S397–S493 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Transporters Stephen PH Alexander1 , Eamonn Kelly2, Alistair Mathie3 ,JohnAPeters4 , Emma L Veale3 , Jane F Armstrong5 , Elena Faccenda5 ,SimonDHarding5 ,AdamJPawson5 , Joanna L -

Effect of Hydrolyzable Tannins on Glucose-Transporter Expression and Their Bioavailability in Pig Small-Intestinal 3D Cell Model
molecules Article Effect of Hydrolyzable Tannins on Glucose-Transporter Expression and Their Bioavailability in Pig Small-Intestinal 3D Cell Model Maksimiljan Brus 1 , Robert Frangež 2, Mario Gorenjak 3 , Petra Kotnik 4,5, Željko Knez 4,5 and Dejan Škorjanc 1,* 1 Faculty of Agriculture and Life Sciences, University of Maribor, Pivola 10, 2311 Hoˇce,Slovenia; [email protected] 2 Veterinary Faculty, Institute of Preclinical Sciences, University of Ljubljana, Gerbiˇceva60, 1000 Ljubljana, Slovenia; [email protected] 3 Center for Human Molecular Genetics and Pharmacogenomics, Faculty of Medicine, University of Maribor, Taborska 8, 2000 Maribor, Slovenia; [email protected] 4 Department of Chemistry, Faculty of Medicine, University of Maribor, Taborska 8, 2000 Maribor, Slovenia; [email protected] (P.K.); [email protected] (Ž.K.) 5 Laboratory for Separation Processes and Product Design, Faculty of Chemistry and Chemical Engineering, University of Maribor, Smetanova 17, 2000 Maribor, Slovenia * Correspondence: [email protected]; Tel.: +386-2-320-90-25 Abstract: Intestinal transepithelial transport of glucose is mediated by glucose transporters, and affects postprandial blood-glucose levels. This study investigates the effect of wood extracts rich in hydrolyzable tannins (HTs) that originated from sweet chestnut (Castanea sativa Mill.) and oak (Quercus petraea) on the expression of glucose transporter genes and the uptake of glucose and HT constituents in a 3D porcine-small-intestine epithelial-cell model. The viability of epithelial cells CLAB and PSI exposed to different HTs was determined using alamarBlue®. qPCR was used to analyze the gene expression of SGLT1, GLUT2, GLUT4, and POLR2A. Glucose uptake was confirmed Citation: Brus, M.; Frangež, R.; by assay, and LC–MS/ MS was used for the analysis of HT bioavailability. -
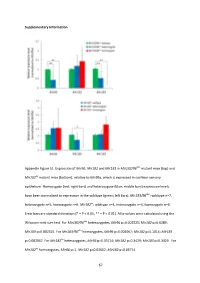
And Mir183 in Mir183/96 Dko Mutant Mice (Top) And
Supplementary Information Appendix Figure S1. Expression of Mir96 , Mir182 and Mir183 in Mir183/96 dko mutant mice (top) and Mir182 ko mutant mice (bottom), relative to Mir99a , which is expressed in cochlear sensory epithelium. Homozygote (red; right bars) and heterozygote (blue; middle bars) expression levels have been normalised to expression in the wildtype (green; left bars). Mir183/96 dko : wildtype n=7, heterozygote n=5, homozygote n=6. Mir182 ko : wildtype n=4, heterozygote n=4, homozygote n=4. Error bars are standard deviation (* = P < 0.05, ** = P < 0.01). All p-values were calculated using the Wilcoxon rank sum test. For Mir183/96 dko heterozygotes, Mir96 p=0.002525; Mir182 p=0.6389; Mir183 p=0.002525. For Mir183/96 dko homozygotes, Mir96 p=0.002067; Mir182 p=0.1014; Mir183 p=0.002067. For Mir182 ko heterozygotes, Mir96 p=0.05714; Mir182 p=0.3429; Mir183 p=0.3429. For Mir182 ko homozygotes, Mir96 p=1; Mir182 p=0.02652; Mir183 p=0.05714. 67 68 Appendix Figure S2. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir183/96 dko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 69 70 Appendix Figure S3. Individual ABR thresholds of wildtype, heterozygous and homozygous Mir182 ko mice at all ages tested. Number of mice of each genotype tested at each age is shown on the threshold plot. 71 Appendix Figure S4. Mean ABR waveforms at 12kHz, shown at 20dB (top) and 50dB (bottom) above threshold (sensation level, SL) ± standard deviation, at four weeks old. -

Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon
Towards the Elucidation of Orphan Lysosomal Transporters Quentin Verdon To cite this version: Quentin Verdon. Towards the Elucidation of Orphan Lysosomal Transporters. Cancer. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS144. tel-01827233 HAL Id: tel-01827233 https://tel.archives-ouvertes.fr/tel-01827233 Submitted on 2 Jul 2018 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS144 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY PREPAREE A L’UNIVERSITE PARIS-SUD ECOLE DOCTORALE N°568 BIOSIGNE | Signalisations et réseaux intégratifs en biologie Spécialité de doctorat : aspects moléculaires et cellulaires de la biologie Par Mr Quentin Verdon Towards the elucidation of orphan lysosomal transporters: several shots on target and one goal Thèse présentée et soutenue à Paris le 29/06/2016 » : Composition du Jury : Mr Le Maire Marc Professeur, Université Paris-Sud Président Mr Birman Serge Directeur de recherche, CNRS Rapporteur Mr Murray James Assistant professor, Trinity college Dublin Rapporteur Mr Goud Bruno Directeur de recherche, CNRS Examinateur Mr Gasnier Bruno Directeur de recherche, CNRS Directeur de thèse Mme Sagné Corinne Chargée de recherche, INSERM Co-directeur de thèse Table of contents Remerciements (acknowledgements) 6 Abbreviations 7 Abstracts 10 Introduction 12 1 Physiology of lysosomes 12 1.1 Discovery and generalities 12 1.2 Degradative function 13 1.3. -

Biomarker Status of the Human Equilibrative Nucleoside
cancers Article “Open Sesame?”: Biomarker Status of the Human Equilibrative Nucleoside Transporter-1 and Molecular Mechanisms Influencing its Expression and Activity in the Uptake and Cytotoxicity of Gemcitabine in Pancreatic Cancer 1,2, 1, 1,3 1 Ornella Randazzo y, Filippo Papini y, Giulia Mantini , Alessandro Gregori , Barbara Parrino 2, Daniel S. K. Liu 4 , Stella Cascioferro 2 , Daniela Carbone 2 , 1,5 4,6, 1,7, Godefridus J. Peters , Adam E. Frampton * , Ingrid Garajova y and Elisa Giovannetti 1,3,* 1 Department of Medical Oncology, Cancer Center Amsterdam, Amsterdam UMC, VU University Medical Center (VUmc), 1081 HV Amsterdam, The Netherlands; [email protected] (O.R.); [email protected] (F.P.); [email protected] (G.M.); [email protected] (A.G.); [email protected] (G.J.P.); [email protected] (I.G.) 2 Dipartimento di Scienze e Tecnologie Biologiche Chimiche e Farmaceutiche (STEBICEF), Università degli Studi di Palermo, 90123 Palermo, Italy; [email protected] (B.P.); [email protected] (S.C.); [email protected] (D.C.) 3 Cancer Pharmacology Lab, AIRC Start Up Unit, Fondazione Pisana per la Scienza, 56017 Pisa, Italy 4 Division of Cancer, Department of Surgery & Cancer, Imperial College, Hammersmith Hospital campus, London W12 0NN, UK;; [email protected] 5 Department of Biochemistry, Medical University of Gdansk, 80-210 Gdansk, Poland 6 Faculty of Health and Medical Sciences, The Leggett Building, University of Surrey, Guildford GU2 7XH, UK 7 Medical Oncology Unit, University Hospital of Parma, Via Gramsci 14, 43126 Parma, Italy * Correspondence: [email protected] (A.E.F.); [email protected] (E.G.); Tel.: +31-003-120-444-2633 (E.G.) These authors contributed equally to this paper.