Supplementary Materials For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Secretariat Phones
H.P.SECRETARIAT TELEPHONE NUMBERS th AS ON 09 August, 2021 DESIGNATION NAME ROOM PHONE PBX PHONE NO OFFICE RESIDENCE HP Sectt Control Room No. E304-A 2622204 459,502 HP Sectt Fax No. 2621154 HP Sectt EPABX No. 2621804 HP Sectt DID Code 2880 COUNCIL OF MINISTERS CHIEF MINISTER Jai Ram Thakur 1st Floor 2625400 600 Ellerslie 2625819 Building 2620979 2627803 2627808 2628381 2627809 FAX 2625011 Oak-Over 858 2621384 2627529 FAX 2625255 While in Delhi Residence Office New Delhi Tel fax 23329678 Himachal Sadan 24105073 24101994 Himachal Bhawan 23321375 Advisor to CM + Dr.R.N.Batta 1 st Floor 2625400 646 2627219 Pr.PS.to CM Ellersile 2625819 94180-83222 PS to Pr.PS to CM Raman Kumar Sharma 2625400 646 2835180 94180-23124 OSD to Chief Minister Mahender Kumar Dharmani E121 2621007 610,643 2628319 94180-28319 PS to OSD Rajinder Verma E120 2621007 710 94593-93774 OSD to Chief Minister Shishu Dharma E16G 2621907 657 2628100 94184-01500 PS to OSD Balak Ram E15G 2621907 757 2830786 78319-80020 Pr. PS to Diwan Negi E102 2627803 799 2812250 Chief Minister 94180-20964 Sr.PS to Chief Minister Subhash Chauhan E101 2625819 785 2835863 98160-35863 Sr.PS to Chief Minister Satinder Kumar E101 2625819 743 2670136 94180-80136 PS to Chief Minister Kaur Singh Thakur E102 2627803 700 2628562 94182-32562 PS to Chief Minister Tulsi Ram Sharma E101 2625400 746,869 2624050 94184-60050 Press Secretary Dr.Rajesh Sharma E104 2620018 699 94180-09893 to C M Addl.SP. Brijesh Sood E105 2627811 859 94180-39449 (CM Security) CEO Rajeev Sharma 23 G & 24 G 538,597 94184-50005 MyGov. -

List of Candidates (Provisionally Eligiblenot Eligible) for Research
Central University of Rajasthan Ph.D. Admissions 2021 List of Candidates (Provisionally Eligible/Not Eligible) for Research Aptitude Test, Presentation and Interview Ph.D. in Atmospheric Sciences Provisionally ID No. Student_Name Eligible Remarks (YES / NO) 116 SUBHASH YADAV YES 1227 ANIRUDH SHARMA YES 1345 ROHITASH YADAV YES 219 RITU BHARGAVA RAI YES Subject to submission of application through proper channel 363 SURINA ROUTRAY YES 396 SHANTANU SINHA YES 404 SHRAVANI BANERJEE YES 915 RAJNI CHOUDHARY YES 956 SONIYA YADAV YES 425 JYOTI PATHAK YES 85 SURUCHI YES 1404 BARKHA KUMAWAT NO No proof of qualifying any National Level Examination 988 ANJANA YADAV NO No proof of National Level Examination NOTE: Grievances, if any, w.r.t. above list may be submitted on [email protected] on or before 11th August 2021. The above list is only to appear for Research Aptitude Test, Presentation and Interview and it does not give any claim of final Admission to Ph.D. programme. Page No. 1 of 31 Central University of Rajasthan Ph.D. Admissions 2021 List of Candidates (Provisionally Eligible/Not Eligible) for Research Aptitude Test, Presentation and Interview Ph.D. in Biochemistry Provisionally ID No. Student_Name Eligible Remarks (YES / NO) 1000 SIMRAN BANSAL YES 1032 RAJNI YES 1047 AKASH CHOUDHARY YES 1048 AKASH CHOUDHARY YES 1050 ADITI SHARMA YES 1065 NAVEEN SONI YES 1168 NIDHI KUMARI YES 1185 ANISHA YES 1206 ABHISHEK RAO YES 1212 RASHMI YES 1228 NEELAM KUMARI YES 1241 ANUKRATI ARYA YES 1319 -

Prof. J. N. Gautam)
Dr. Jagdish Narain Gautam School of Studies in Library and Information Science, Jiwaji University, Gwalior – 474011. Mob. +919425364178 PHONE NO.0751-2343176 (R) 0751-2442627 E-mail: [email protected] FATHER'S NAME : Late Shri Asha Ram Gautam SPECIALIZATION : Knowledge Organization and Processing (Classification & Cataloguing), Library Information and Society, Library Management System and Library Automation and Networking. PRESENT POSITION : Professor, SOS, Library and Information Science. Rector, Jiwaji University, Gwalior. OTHER RESPONSIBILITIES : : Dean Faculty of Arts, Since 2006 : Chairman Board of Studies in LIS, Since 1996 : Member of Academic Council : Member of Planning and Evaluation Board : Chief Warden since 2005-2009 ACADEMIC EXPERIENCE : 22 Years QUALIFICATIONS : Degree Year Board/University Subject/Title Division Ph.D. 1991 Jiwaji University, The Information needs and Information Gwalior seeking of Physics & Chemistry Teachers in University and Collages of Gwalior and Chambal Division – A Scientific Study M.Lib.I.Sc 1986 Dr. H. S. Gour Library and Information Science II University, Sagar M.Sc. (Phy. 1984 Dr. H. S. Gour Anthropology I Anthro.) University, Sagar PROFESSIONAL EXPERIENCE: 22 Years (As per the details below) S. No. Post Held Department/Institution Duration 1. Librarian (Part -Time) Lok Kala Academy, Sagar. 01.09.1984 to 01.08.1985 2. Librarian Govt. P.G. College, Tikamgarh. 03.08.1985 to 04.09.1985 3. Librarian Govt. Degree College, Sitapur. 04.10.1986 to 31.07.1987 4. Assistant Librarian Central Library, Jiwaji University, 20.11.1987 to 08.05.1996 Gwalior. 5. In charge University Central Library, Jiwaji University, 09.05.1996 to 31.03.2010 Librarian Gwalior. TEACHING EXPERIENCE : 22 Years (As per the details below) B.Lib.I.Sc: Papers Taught - Knowledge organization and Processing (Classification & Cataloguing Theory) - Knowledge organization and Processing (Classification Practical) CC Rev. -
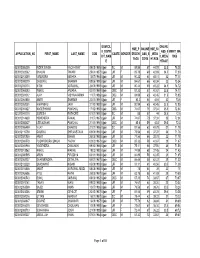
Application No First Name Last Name Dob Domicil
DOMICIL ONLINE HSE_P ONLINE HSE_50 E_DISTR _ASS_5 MERIT_MA APPLICATION_NO FIRST_NAME LAST_NAME DOB CASTE GENDER ERCEN _ASS_S _WEIG ICT_NAM 0_WEIG RKS TAGE CORE HTAGE E HTAGE 030812056209 INDER SINGH KACHHWAY 09/05/1986 Ujjain SC M 89.56 67 44.78 33.5 78.28 030812023062 SHALINI TIWARI 29/04/1987 Ujjain UR F 85.78 69 42.89 34.5 77.39 030812013389 VIRENDRA MISHRA 13/07/1984 Ujjain UR M 90.22 64 45.11 32 77.11 030812093079 DHEERAJ SHARMA 08/06/1990 Ujjain UR M 84.67 66 42.34 33 75.34 030812032174 NITIN AGRAWAL 26/09/1989 Ujjain UR M 80.44 69 40.22 34.5 74.72 030812068283 RAHUL ANJANA 02/11/1989 Ujjain OBC M 81.33 67 40.67 33.5 74.17 030812001012 AJAY VISHWAKARMA 11/07/1986 Ujjain OBC M 84.89 63 42.45 31.5 73.95 030812061800 AARTI SHARMA 22/01/1991 Ujjain UR F 83.2 64 41.6 32 73.6 030812053051 HIMANSHU JAIN 31/10/1988 Ujjain UR M 80.89 65 40.45 32.5 72.95 030312081427 NAGESHWAR PANCHAL 17/12/1989 Ujjain OBC M 74.67 71 37.34 35.5 72.84 030312041110 SURESH RATHORE 01/07/1991 Ujjain SC M 88 57 44 28.5 72.5 030812014630 HEMENDRA RAWAL 31/07/1987 Ujjain UR M 74.67 70 37.34 35 72.34 030812055427 LEELADHAR PANCHAL 21/11/1991 Ujjain OBC M 85.6 59 42.8 29.5 72.3 030812086499 VIJAY DHAKITE 21/01/1986 Ujjain SC M 81.56 62 40.78 31 71.78 030812115790 GAURAV SHRIVASTAVA 08/06/1986 Ujjain UR M 75.56 68 37.78 34 71.78 030812020950 ANKIT BIHANI 25/05/1988 Ujjain UR M 77.56 66 38.78 33 71.78 030812069570 PUSHPENDRA SINGH YADAV 02/03/1987 Ujjain OBC M 85.33 58 42.67 29 71.67 030812041960 YOGENDRA CHAUHAN 09/10/1986 Ujjain UR M 75.11 68 37.56 34 71.56 030812013862 -

Historical Perspective of Rajput Society
SOCIO - HISTORICAL PERSPECTIVE OF RAJPUT SOCIETY DR. YASHPALSINH V. RATHOD At. Dhamboliya Dist. Arvvali (GJ) INDIA Maharaja Shree Ranjorsinh has identified Kshatriya Pedigree, as per him 16 from Sun, 4 from Moon, 2 from Nagvanshi, 3 from Rushivanshi and 11 from Agnivanshi are also Kshatriya. There are some assumptions for 36 Descent. But in sun vansh Rathod, Katchvaha, Sisodiya, Badgujar,Kathariya, Sikarval, Nikumbh and Rekvar are considered. European historian Easterson, Indian veda and cultural tradion indicates Rajputs are ancient Aaryajati. These gens are from Rajput and they had ruled India since vaidik period. That cast has provided bravely protection to our country, religion and culture. INTRODUCTION This article shows origin of Rajput society and their social, cultural, geographical, Economical and historical aspects. People from same cast who are living in different areas of Gujarat should know their Ancestor, their detail information is necessary today. Today’s young generation does not have proper information regarding their Descent.they should know their gotra and sub cast properly. They don’t believe in relation of brotherhood. If we ask question to any rauput youngster regarding their Descent and sub branch, that answer is not satisfactory. That’s why origin and historical detail study of Rathod Descent is required. Here detail information written of 36 Descent and their sub branch indicated in this article. Rathod Descent community is in majority in some areas of Gujarat, also in some part of the state there are Solanki, Chauhan, Parmar, Zala,Waghela etc… community are living. Kulgury, Bhat, Charan, Vahivarcha, Rani, Maga, Rao, Dhol community has played an important role in Indian Kshatriya gaurav gatha. -

Hindu Tribes and Castes As Represented in Benaras
HINDU TRIBES AND CASTES AS REPRESENTED IN BENARAS WITH ILLUSTRATIONS M. A. SHERRING ASIAN EDUCATIONAL SERVICES NEW DELHI * CHENNAI * 2008 CONTENTS. Page Introduction ... ... ... ... ... -... ... ... ... i—ix PART L—THE BRAHMANICAL TRIBES. CHAPTER I. The Brahman in relation to the Past, the Present, and the Future ... ... ... ... 1 CHAPTER II. Genealogy of the Brahmanical Tribes, and the Classification of their Orders according to their observ- ance of the Vedic Rituals—Honorary Titles—Divisions into Clans—Six Special Duties—Religious Ceremonies of the Brahmane—The Nakshatras ... ... ... ... ... ... 6 CHAPTER III. Ten Principal Brahmanical Tribes. Supplementary Tribes. The Five Gaur Tribes of Northern India. The First Tribe—Kanyakubja Brahmane. First Sub-tribe—The Kanyakubja Brahmane Proper. Gotras. Clans. Kanoujiya Brahmane of Bengal—Varendra, Rarhiya, Pashchatiya, and Daksh- inatiya ··· ... ··· ·«· ··· ·•· ··· .·· ··. ·»· 19 CHAPTER IV. The Sarjuparia or Sarwariya Brahmane. The Sawalakhî Brahmane. The Maha-Brahman, or Acharja. The Ganga-putra, or Son of the Gange«. The Gayâwâl The PryâgwâL The Ojha. The Bhanreriya ... ... ·*· ... ... *·· ... .*· ... ... 29 CHAPTER V. The Bhûinhâr Brahman«. The Kinwars. The Bemwars. The Sakarwars. The Dunwârs. The Kast- wars. Historical Sketches of the Families of the Maharaja of Benares, the late Raja Sir Deo Narain Singh Bahadur, and Baba Futteh Narain Singh ... ... ... ... ... 39 CHAPTER VL The Jijhotiya Brahmans. Gotras and Clans ... ·.· ... ... ... ... ... '55 CHAPTER VII. The Sanâdhiya Brahmans. Gotras and Titles ... ... ... ... ... ... ... 57 CHAPTER VII. The Second Tribe of Gaur Brahmans—Sâraswat. Their Antiquity. Their original Home. Four great Divisions—Panjâtî, Ashtbans, Barahî, and Bawan or Bhunjahi ... ... ,„ ... 62 vi CONTENTS. Page CHAPTER IX. The Third Tribe of Gaur Brahmans—Gaur. The Taga Brahmans. Their Origin. Sub-divisions. Clans ... ... ... ... ... ... ... ... ... ... ··· 66 CHAPTER X. -

List of Officers Who Attended Courses at NCRB
List of officers who attened courses at NCRB Sr.No State/Organisation Name Rank YEAR 2000 SQL & RDBMS (INGRES) From 03/04/2000 to 20/04/2000 1 Andhra Pradesh Shri P. GOPALAKRISHNAMURTHY SI 2 Andhra Pradesh Shri P. MURALI KRISHNA INSPECTOR 3 Assam Shri AMULYA KUMAR DEKA SI 4 Delhi Shri SANDEEP KUMAR ASI 5 Gujarat Shri KALPESH DHIRAJLAL BHATT PWSI 6 Gujarat Shri SHRIDHAR NATVARRAO THAKARE PWSI 7 Jammu & Kashmir Shri TAHIR AHMED SI 8 Jammu & Kashmir Shri VIJAY KUMAR SI 9 Maharashtra Shri ABHIMAN SARKAR HEAD CONSTABLE 10 Maharashtra Shri MODAK YASHWANT MOHANIRAJ INSPECTOR 11 Mizoram Shri C. LALCHHUANKIMA ASI 12 Mizoram Shri F. RAMNGHAKLIANA ASI 13 Mizoram Shri MS. LALNUNTHARI HMAR ASI 14 Mizoram Shri R. ROTLUANGA ASI 15 Punjab Shri GURDEV SINGH INSPECTOR 16 Punjab Shri SUKHCHAIN SINGH SI 17 Tamil Nadu Shri JERALD ALEXANDER SI 18 Tamil Nadu Shri S. CHARLES SI 19 Tamil Nadu Shri SMT. C. KALAVATHEY INSPECTOR 20 Uttar Pradesh Shri INDU BHUSHAN NAUTIYAL SI 21 Uttar Pradesh Shri OM PRAKASH ARYA INSPECTOR 22 West Bengal Shri PARTHA PRATIM GUHA ASI 23 West Bengal Shri PURNA CHANDRA DUTTA ASI PC OPERATION & OFFICE AUTOMATION From 01/05/2000 to 12/05/2000 1 Andhra Pradesh Shri LALSAHEB BANDANAPUDI DY.SP 2 Andhra Pradesh Shri V. RUDRA KUMAR DY.SP 3 Border Security Force Shri ASHOK ARJUN PATIL DY.COMDT. 4 Border Security Force Shri DANIEL ADHIKARI DY.COMDT. 5 Border Security Force Shri DR. VINAYA BHARATI CMO 6 CISF Shri JISHNU PRASANNA MUKHERJEE ASST.COMDT. 7 CISF Shri K.K. SHARMA ASST.COMDT. -

Prof. Madhu Rajput Department of Western History, Lucknow University
Profile: Prof. Madhu Rajput Department of Western History, Lucknow University. Prof. Madhu Rajput is a faculty in the Department of Western History, University of Lucknow. Her field of specialization is ‘Women Studies’ and ‘South Asia’. She has extensively traveled in Bhutan and India and researched on women in these countries and has completed 03 Major Projects on ‘Bhutanese Women’, ‘Bhutanese State, Law and People’, and ‘Tibetan Women Living in Exile in India’. She has authored 08 books on Bhutan and North East India and has published 34 papers in various national journals. 07 of her articles have also been published in ‘Bhutan Times’ and ‘Tibetan Review’. She has attended and presented her papers in 16 National and International seminars and has organized 03 International seminars in her own University. She has conducted Workshops and lectures in Bhutan and various parts of India on women studies. A member of reputed institutions such as Indian History Congress, Indian Social Science Congress, and U.P. History Congress, Prof. Rajput is also an examiner in 08 Public Service Commissions and Universities. She conducts Refresher Courses as an expert for the College and University lecturers. She has guided 08 Ph.Ds. For her significant contribution in social studies, she has been awarded the ‘Uttar Pradesh Ratan Award’ by Shri H.N. Dixit, Hon’ble Speaker, U.P. Legislative Assembly on 23 September 2018, and ‘Shakti Samman’, on 08 March 2016 in the field of education, by Social Active Welfare Trust and ‘Sanskritiki’ Lucknow University, Uttar Pradesh. She is active in social service through conducting welfare programs such as free counselling sessions and career guidance to students. -

A Brief Profile of DR. U. S. GAUTAM
A Brief Profile of DR. U. S. GAUTAM Dr U S Gautam is Director, ICAR - Agricultural Technology Application Research Institute (ATARI), Rawatpur, Kanpur, (UP). Born on January 23, 1963 in village Mirzamurad, Varanasi district Uttar Pradesh, Dr. U.S. Gautam completed schooling while helping family farming, graduate and post graduate from Institute of Agricultural Science B.H.U., Varanasi in 1986, with specialization in Extension Education and earned Ph.D with ICAR fellowship in Dairy Extension from NDRI, Karnal in November, 1989 with specialization of Decision Making Component of Dairy Farmers for Better Adoption of Dairy Practices. Began his Professional Career as Assistant Extension Specialist (Agriculture Extension) in KVK,RRS, Bajoura, Kullu, from Himachal Pradesh Krishi Vishwa Vidhyalaya, in November, 1989, and he was selected as Senior Scientist in (1998),Principal Scientist & then Head, Socio Economic and Extension Research Programme (2001 to 2004) at ICAR Research Complex for Eastern Region, Patna, Bihar. He completed one tenure as Zonal Project Director, ZPD, Zone VII, ICAR, JNKVV Campus, Adhartal Jabalpur, (MP) from 30-6-2006 to 03-03- 2012, ZPD, Zone VII is comprises of three States i.e. MP, Chhattisgarh and Orissa for monitoring and coordinating with 100 KVKs of biggest Zone in Country. The charge resume as Director ,ICAR-ATARI on 24th February,2015,Which is comprises of two States i.e. Utter Pradesh(UP) & Uttrakhand (UK) for monitoring and coordinating of 81 KVKs in Country. Dr. Gautam guided two Ph.D students and ten M.Sc Students thesis evaluated. Dr. Gautam was recipient of several National/International Awards; among them some of important are “Swamy Sahajanand Saraswati Extension Scientist Award and ICAR Award for Outstanding Multidisciplinary Team Research in Agricultural and Allied Science” as a team leader. -

HISTORY ANCIENT PERIOD in the Epic Period, the Region Covered By
HISTORY ANCIENT PERIOD In the epic period, the region covered by the present district of Gorakhpur, known as Karapatha, which formed part of the kingdom of Kosala, an important centre of Aryan culture and civilization. The renowned ascetic Gorakh Nath gave name and fame to this district by practicing austerities on the spot where the famous temple named after him stands. It appears that the earliest known monarch ruling over this region with his capital at Ayodhya, was Iksvaku, who founded the solar dynasty of Ksatriyas. It produced a number of illustrious kings till the accession of Ram, who was the greatest ruler of this dynasty. Ram had divided the kingdom, during his lifetime, into small principalities. He coronated his eldest son Kusa as the king of Kusavati present Kushinagar which lay in the Gorakhpur district till 1946. After Ram's renunciation of the world Kusa left Kusawati (Kushinagar) and repaired to Ayodhya. His cousin, Chandraketu, son of Lakshmana, even the epithet of malla (valiant) in the Ramayan, thereupon took possession of this region. The Mahabharata mentions that at the Rajasuya (imperial) sacrifice performed by Yudhisthira, a behest to conquer the east was given to Bhimasena who in turn subjugated the principality of Gopalak (identified with Gopalpur of Bansgaon tahsil). Close to Gopalpur at Bhimtola, Bhimasena is said to have reposed after his victory. The discovery of a series of enormous mounds at Gopalpur and around it suggests that the places has been old sites of extensive cities. A few development in the political history of the district during the post- Mahabharata period is the functioning of a number of republics under the sovereignty of the kingdom of Kosala. -

Sudden Death of Sher Shah Suri Humayun Re
GAUTAM SINGH UPSC STUDY MATERIAL – INDIAN HISTORY 0 7830294949 UNIT 45 – UPSC -Humayun recovers the throne of Delhi India's History : Medieval India : Humayun recovers the throne of Delhi - 1555 Sudden death of Sher Shah Suri In 1545, after the accidental death ofSher Shah, his son Jalal Khan succeeded him. Jalal Khan got the title of Islam Shah, commonly known as Salim Shah. Islam Shah was as capable as his father and kept his father's kingdom intact. He followed his father's reforms and kept the army intact. Unfortunately, he ruled for only nine years. Following his death in November 1554, disorder followed. His minor son, Firuz Khan, was murdered by his maternal uncle, Mubariz Khan, and there was total confusion in the empire. Mubariz Khan took the throne and assumed the title of Muhammad Adil Shah. Adil Shah was a worthless ruler. He left the affairs of the kingdom in the hands of his chief minister Hemu. Hemu was a capable man but his ambition to seize the throne did not draw his attention towards the disintegration of the kingdom. There were revolts in various parts. Sher Shah's nephew Sikander Sur declared himself independent in the Punjab. Humayun re-conquers Delhi This conflicting situation encouraged Humayun to make an attempt to restore the lost empire after about fifteen years. He got an army of 14,000 men from Persia and succeeded in conquering Kabul and Kandhahar with the help of Shah of Iran in 1545. In November 1554, he marched to reconquer Hindustan, for which he got an excellent opportunity in the civil wars among the surs. -

College of Vocational Studies University of Delhi Internal Assessment Marks
COLLEGE OF VOCATIONAL STUDIES UNIVERSITY OF DELHI INTERNAL ASSESSMENT MARKS - MAY-2018 B.A. (VOCATIONAL STUDIES) SME - IV SEMESTER Sr. College Paper Max Obt. Student Name Exam Rollno Sem Paper Name Signature No. Rollno Code Marks Marks 1 AADESH 661 16013576001 IV 62031412 Advanced English: Stream A 25 14 AADESH 661 16013576001 IV 62153401 Business Communications and personality Development 25 19 AADESH 661 16013576001 IV 62154406 MSMEs Policy Framework 25 0 AADESH 661 16013576001 IV 62154413 Labour and Development in India 25 19 2 AKASH AWANA 668 16013576004 IV 62031412 Advanced English: Stream A 25 15 AKASH AWANA 668 16013576004 IV 62153401 Business Communications and personality Development 25 15 AKASH AWANA 668 16013576004 IV 62154406 MSMEs Policy Framework 25 9 AKASH AWANA 668 16013576004 IV 62154413 Labour and Development in India 25 14 3 ALISHA ANSARI 670 16013576005 IV 62031412 Advanced English: Stream A 25 17 ALISHA ANSARI 670 16013576005 IV 62153401 Business Communications and personality Development 25 22 ALISHA ANSARI 670 16013576005 IV 62154406 MSMEs Policy Framework 25 17 ALISHA ANSARI 670 16013576005 IV 62154413 Labour and Development in India 25 21 4 AMAN GOEL 603 16013576006 IV 62031412 Advanced English: Stream A 25 20 AMAN GOEL 603 16013576006 IV 62153401 Business Communications and personality Development 25 23 AMAN GOEL 603 16013576006 IV 62154406 MSMEs Policy Framework 25 23 AMAN GOEL 603 16013576006 IV 62154413 Labour and Development in India 25 24 5 BHARAT GUPTA 601 16013576008 IV 62031412 Advanced English: Stream