Migration and Differentiation of Murine Germinal Center Derived B Cell Subsets in the Course of the NP- Specific Immune Response
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies of Endothelial and Leukocyte Cell Adhesion Molecules in Renal Transplantation
ì1-3-r\"1\, Studies of Endothelial and Leukocyte Cell Adhesion Molecules in Renal Transplantation A Thesis submitted to the University of Adelaide as the requirement for the degree of Doctor of PhilosoPhy by Warwick L Grooby BSc (Hons) Department of Medicine, University of Adelaide and Transplantation Immunology Laboratory The Queen Elizabeth Hospital November \996 I TABLE OF CONTENTS Table of Contents I Summary vlll Declaration x Acknowledgments xi Dedication xii Publications and Presentations xtll Abbreviations xv CHAPTER 1 Introduction 1..L Introduction 2 L.2 Cell Adhesion Molecules 3 1,.2.1, Integrin Family J 8 1.2.1,.1. PL integrin subfamilY 'J,.2.1..2 9 P2 integrin subfamilY 11 1,.2.1.3 P7 integrin subfamilY 1,.2.2 Immunoglobulin (Ig) SuperfamilY 12 1,.2.2.1 ICAM-L (Intercellular adhesion molecule-l) 14 t.2.2.2 ICAM-2 (Letercellular adhesion molecule-2) 15 1..2.2.3 ICAM-3 (Intercellular adhesion molecule-3) 15 t.2.2.4 VCAM-L (Vascular cell adhesion molecule-1) 16 1,.2.2.5 PECAM-L (Platelet-endothelial cell adhesion molecule-1') 18 L.2.2.6 MAdCAM-L (Mucosal addressin cell adhesion molecule-L) 19 1,.2.3 Selectin Family 19 1,.2.3.1 E-selectin 20 1,.2.3.2 P-selectin 22 7.2.3.3 L-selectin 23 1.2.4 Vascular Mucins (addressins) 24 L.2.4.L GIyCAM-1 (Glycosylation-dependent cell adhesion molecule-1) 24 1,.2.4.2 CD34 25 1,.2.5 Cadherins 25 1..2.6 CD4O 26 1.2.7 CDM 27 l1 L.3 Vascular Endothelium 28 L.3.1 MorpholoW of Endothelial Cells 29 1.3.1.1 High Endothelial Venules 30 1.3.2 Study of Endothelial Cells in aitro 31 L.3.2J, Large Vessel Endothelial Cells 31 1.3.2.2 Microvascular Endothelial Cells 32 1,.3.2.3 Transformed Endothelial Cells 33 1.3.3 Heterogeneity of Vascular Endothelium 33 1.3.3.1 Weibel-Palade bodies (WPBs) 33 1,.3.3.2 von Willebrand factor (vWF) 34 1.3.3.3 Biosynthetic Heterogeneity 34 L.3.3.4 Antigenic HeterogeneitY 35 1..3.4 Endothelial Cell Functions 36 1..3.4.1. -

C07k - 2021.08
CPC - C07K - 2021.08 C07K PEPTIDES (peptides in foodstuffs A23; obtaining protein compositions for foodstuffs, working-up proteins for foodstuffs A23J; preparations for medicinal purposes A61K; peptides containing beta-lactam rings C07D; cyclic dipeptides not having in their molecule any other peptide link than those which form their ring, e.g. piperazine-2,5-diones, C07D; ergot alkaloids of the cyclic peptide type C07D 519/02; macromolecular compounds having statistically distributed amino acid units in their molecules, i.e. when the preparation does not provide for a specific; but for a random sequence of the amino acid units, homopolyamides and block copolyamides derived from amino acids C08G 69/00; macromolecular products derived from proteins C08H 1/00; preparation of glue or gelatine C09H; single cell proteins, enzymes C12N; genetic engineering processes for obtaining peptides C12N 15/00; compositions for measuring or testing processes involving enzymes C12Q; investigation or analysis of biological material G01N 33/00) Relationships with other classification places An amino acid per se is classified in C07D while peptides (starting from dipeptides) are classified in C07K. Subclass C07K is a function oriented entry for the compounds themselves and does not cover the application or use of the compounds under the subclass definition. For classifying such information other entries exist, for example: preservation of bodies of humans or animals or plants or parts thereof; Biocides, e.g. as disinfectants, as pesticides, as herbicides; pest repellants or attractants; plant growth regulators are classified in A01N. Preparations for medical, dental, or toilet purposes are classified in A61K. Amino acids or derivatives thereof are classified in C07C or C07D. -

Flow Cytometry
Flow cytometry 謝長奇 中西醫結合研究所 流式細胞儀應用 A brief list of applications that use flow cytometers includes: Disease diagnosis Chromosome karyotyping Cell function analysis Cancer therapy monitoring Detecting fetal cells Cell kinetics Identifying tumor cells Cytogenetics Fundamental cell biology 1 流式細胞儀應用 免疫細胞分析 Cell surface marker analysis Intracellular cytokines analysis Platelet analysis 細胞凋亡 上樣與資料分析 免疫細胞表面標記分析 2 參與免疫反應的細胞 參與免疫反應的細胞 Primary organs bone marrow thymus Secondary organs and tissues spleen lymph nodes Peyer's patches 3 參與免疫反應的組織 lymph node spleen LYMPHOCYTE SURFACE ANTIGENS IMMUNOGLOBULINS: "Antibodies" secreted by or found on B-Cells. Has a huge range of specificities achieved by DNA rearrangement. Five general types (see below): IgG, IgA, IgE, IgD, IgM 4 LYMPHOCYTE SURFACE ANTIGENS T-CELL (TcR) RECEPTORS: They bind to the Antigen-Presenting Cell. VARIABLE REGIONS are on the T-Cell Receptor. They allow us to develop variability and diversity in the immune response. "CD" ANTIGENS: Systematic classification of surface-antigens with diverse functions. Cell-surface markers. Cluster of Differentiation (CD) Antigens Leukocytes express distinct assortments of molecules on their cell surfaces many of which reflect either different stages of their lineage-specific differentiation different states of activation or inactivation 5 Cell surface immunophenotypes different leukocyte subpopulations, including the functionally distinct mature lymphocyte subpopulations B-cells helper T-cells (TH) cytotoxic T-cells (TC) natural killer (NK) cells Antigen markers on mature lymphocyte populations 6 Conventions for Naming Leukocyte Surface Molecules Named according to a particular function affected by an anti-leukocyte mAb the lymphocyte function-associated antigen 1, or LFA-1, was so named because antibodies recognizing this structure interfere with lymphocyte cell adhesion events and optimal lymphocyte function. -

Datasheet: MCA1582A488 Product Details
Datasheet: MCA1582A488 Description: MOUSE ANTI HUMAN CD83:Alexa Fluor® 488 Specificity: CD83 Other names: HB15 Format: ALEXA FLUOR® 488 Product Type: Monoclonal Antibody Clone: HB15e Isotype: IgG1 Quantity: 100 TESTS/1ml Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Flow Cytometry Neat Where this antibody has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Suggested working dilutions are given as a guide only. It is recommended that the user titrates the antibody for use in their own system using appropriate negative/positive controls. Target Species Human Species Cross Reacts with: Cynomolgus monkey, Chimpanzee, Baboon, Rhesus Monkey, Tasmanian Devil Reactivity N.B. Antibody reactivity and working conditions may vary between species. Product Form Purified IgG conjugated to Alexa Fluor® 488 liquid Max Ex/Em Fluorophore Excitation (nm) Emission (nm) Alexa Fluor®488 495 519 Preparation Purified IgG prepared by affinity chromatography on Protein G from tissue culture supernatant Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide Stabilisers 1% Bovine Serum Albumin Approx. Protein IgG concentration 0.05 mg/ml Concentrations Immunogen Cos cells transfected with HB15 cDNA. Page 1 of 4 External Database Links UniProt: Q01151 Related reagents Entrez Gene: 9308 CD83 Related reagents Fusion Partners Spleen cells from immunised BALB/c mice were fused with cells of the mouse NS1 myeloma cell line. -

The Role of ATG16L1 in Chronic Inflammatory Bowel Disease
The role of ATG16L1 in chronic inflammatory bowel disease Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Christian-Albrechts-Universität zu Kiel vorgelegt von: Janne Böck Kiel, 2012 Referent/in: Prof. Dr. Rosenstiel Korreferent/in: Prof. Dr. Dr. Bosch Tag der mündlichen Prüfung: 26.06.2012 Zum Druck genehmigt: 02.07.2012 gez. Prof. Dr. Lutz Kipp (Dekan) Table of contents 1 INTRODUCTION ......................................................................................... 1 1.1 The role of autophagy in cell homeostasis and inflammation ....................................... 1 1.2 The role of autophagy in endoplasmic reticulum stress ................................................ 4 1.3 The autophagy protein ATG16L1 ...................................................................................... 6 1.3.1 The role of ATG16L1 in Paneth cell vesicle export ...................................................... 7 1.3.1.1 Paneth cells ......................................................................................................... 7 1.3.1.2 ATG16L1 and Paneth cell vesicle export ............................................................ 8 1.4 Inflammatory bowel disease .............................................................................................. 9 1.4.1 Genetics in inflammatory bowel disease .................................................................... 10 1.4.2 The role of ATG16L1 in Crohn disease .................................................................... -

Pan-Cancer Immunogenomic Analyses Reveals Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade
bioRxiv preprint doi: https://doi.org/10.1101/056101; this version posted May 30, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Pan-cancer immunogenomic analyses reveals genotype-immunophenotype relationships and predictors of response to checkpoint blockade Pornpimol Charoentong†, Francesca Finotello†, Mihaela Angelova†, Clemens Mayer, Mirjana Efremova, Dietmar Rieder, Hubert Hackl, Zlatko Trajanoski* 1Biocenter, Division of Bioinformatics, Medical University of Innsbruck, Innsbruck, Austria †Equal contribution *To whom correspondence should be addressed: [email protected] Zlatko Trajanoski, PhD Biocenter, Division of Bioinformatics Medical University of Innsbruck Innrain 80, 6020 Innsbruck Austria Email: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/056101; this version posted May 30, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. ABSTRACT: Current major challenges in cancer immunotherapy include identification of patients likely to respond to therapy and development of strategies to treat non-responders. To address these problems and facilitate understanding of the tumor-immune cell interactions we inferred the cellular composition and functional orientation of immune infiltrates, and characterized tumor antigens in 19 solid cancers from The Cancer Genome Atlas (TCGA). Decomposition of immune infiltrates revealed prognostic cellular profiles for distinct cancers, and showed that the tumor genotypes determine immunophenotypes and tumor escape mechanisms. -

Functions of Cd45 in Tcr Signaling in Cd4+Cds+ Double-Positive Thymocytes
FUNCTIONS OF CD45 IN TCR SIGNALING IN CD4+CDS+ DOUBLE-POSITIVE THYMOCYTES Gordon W. Cheng A thesis submitted in conformity with the requirements for the degree of Master's of Science Graduate Department of Immunology University of Toronto O Copyright by Gordon W. Cheng (1997) Acquisitions and Acquisitions et Bibliographie Services semices bibliographiques 395 Wellington Street 395, rue Wellington Onawa ON KIA ON4 Onawa ON K1A ON4 Canada Canada Your fib Volro re:erence Our Ida Norre iefeisnce The author has granted a non- L'auteur a accordé une licence non exclusive licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sel1 reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de rnicrofiche/film, de reproduction sur papier ou sur format électronique. The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be printed or othenvise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation. - a Thymocytes Master's of Science (1997) Gordon W. Cheng Graduate Department of Immunology University of Toronto Abstract T ceIl receptor (TCR) signals are essential for normal T cell developrnent, and CD45 is thought 10 be essential for coupling TCR to the intracellular signaling rnachinery. However, T cell development is only partially compromised in CD45-deficient mice. -

CD Antigen Expression (Myeloid & Lymphoid)
CD Antigen Expression Immunological Excellence MYELOID Pluripotent LYMPHOID Stem Cell CD90* CD123 CFU-GEMM Myeloid CD110* CD135* Lymphoid Stem Cell CD117 CD243 Markers expressed CD10 CD117 Stem Cell CD34 CD124 by the majority of CD33 CD112* CD133* CD227* CD38 CD127 CD34 CD117 CD173* CD228 CD90* CD228 Leucocytes CD110* CD123 CD174* CD11a CD55 CD132 CD111* CDw131 CD176 CD18 CD58 CD148 CD29 CD59 CD156b CD44 CD62L CDw217 Pro-B Cell Subcortical Thymocyte CD45 CD82 CD220 CD19 CD79a CD2 CD8 CD46 CD100 CD221 CD22 CD79b CD4 CD38 CD47 CD118 CD222 CD24 CD124 CD5 CD200 CD47R CD120a CD224 CD34 CD127 CD7 CD50 CD120b CD225 BFU-E CFU-GM CD38 CD179a CD53 CD126 CD230 CD33 CD72 CD179b CD54 CD130 CD232 CD34 Myelomonocytic CDw123 Stem Cell Cortical Thymocyte CDw131 CD1a CD4 CD52 CD13 CD111 CD116 Pre-Pre-B Cell CD1b CD5 CD165 CD33 CD112 CDw123 CD10 CD72 THYMUS CD1c CD7 CD200 CD34 CD115 CDw131 CD19 CD79a CD2 CD8 CD20 CD79b CD3 CD38 CD22 CD124 CD24 CD179a CD34 CD179b CFU-E CFU-Meg CFU-M CFU-G CFU-Eo CFU-Bas Pre-B Cell Medullary Medullary CD36 CD34 CD13 CD115 CD13 CD112 CD13 CD34 CD9 CD72 CDw123 CDw123 CD15 CD116 CD15 CD116 CD33 CDw123 CD10 CD74 Thymocyte Thymocyte CDw131 CD110 CD33 CDw123 CD33 CDw123 CD34 CD19 CD79a CD4+ CD8+ CD175s CD111 CDw131 CD111 CDw131 CD116 CD20 CD79b CD112 CDw123 CD22 CD124 CDw131 CD24 CD179a CD38 CD179b BONE MARROW CD40 CD2 CD7 CD150 (CD49f) CD121a Early B Cell CD3 CD27 CD155 CD52 CD127 Reticulocyte Megakaryocyte Promonocyte Myelocyte Myelocyte Myelocyte CD5 CD28 CD48 CD69 CD165 CD41 (CD51) CD13 CD112 CD13 CD114 CD11b CD35 (CD15*) CD19 -
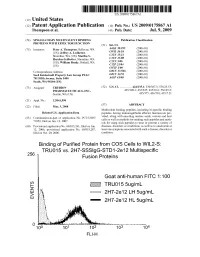
2 IE Truo155ug/Ml G Eo 2H7-2E 12 LH 5Ug/Ml 2H7-2E 12 HL 5Ug/Ml Patent Application Publication Jul
US 20090175867A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0175867 A1 Thompson et al. (43) Pub. Date: Jul. 9, 2009 (54) SINGLE-CHAIN MULTIVALENT BINDING Publication Classification PROTEINS WITH EFFECTOR FUNCTION (51) Int. Cl. (75) Inventors: Peter A. Thompson, Bellevue, WA g Bis CR (US); Jeffrey A. Ledbetter, ( .01) Shoreline, WA (US); Martha S. EN fo C Hayden-Ledbetter, Shoreline, WA CI2N 5/06 :08: S.US); WilliamIIIlam Bradv,5rady, Bothell,Sounell, WA CI2P 2L/04 (2006.01) C7H I/00 (2006.01) Correspondence Address: GOIN 33/566 (2006.01) Seed Intelectuall Property Law Group PLLC GOIN 33/53 (2006.01) 701 Fifth Avenue, Suite 5400 A6IP 43/00 (2006.01) Seatle, WA 98104 (US) (73) Assignee: TRUBION (52) U.S. Cl. ................ 424/135.1; 530/387.3; 536/23.53; PHARMACEUTICALS, INC. 435/320.1; 435/325; 435/69.6; 536/25.3: Seattle, WA (US) s 435/375; 436/501; 435/7.21 (21) Appl. No.: 12/041590 (22) Filed: Mar. 3, 2008 (57)57 ABSTRACT Multivalent binding peptides, including bi-specific binding Related U.S. Application Data peptides, having immunoglobulin effector function are pro (63) Continuation-in-part of application No. PCT/US07/ vided, along with encoding nucleic acids, vectors and host 71052, filed on Jun. 12, 2007. cells as well as methods for making Such peptides and meth s s ods for using Such peptides to treat or prevent a variety of (60) Provisional application No. 60/813.261, filed on Jun. diseases, disorders or conditions, as well as to ameliorate at 12, 2006, provisional application No. -

Immunological Significance of Mycobacterium Leprae Cell Walls
Proc. Nadl. Acad. Sci. USA Vol. 85, pp. 1917-1921, March 1988 Immunology Immunological significance of Mycobacterium leprae cell walls (cell-mediated immunity/delayed-type hypersensitivty/peptidoglycan-protein complex/leprosy/precursor frequency analysis) JOHANNE MELANCON-KAPLAN*, SHIRLEY Wu HUNTERt, MICHAEL MCNEILt, CAROL STEWARTt, ROBERT L. MODLINf, THOMAS H. REAt, JACINTO CONVIT§, PADMINI SALGAME*, VIJAY MEHRA*, BARRY R. BLOOM*, AND PATRICK J. BRENNANt¶ *Department of Microbiology and Immunology, Albert Einstein College of Medicine, Yeshiva University, Bronx, NY 10461; tDepartment of Microbiology, Colorado State University, Fort Collins, CO 80523; tSection of Dermatology, University of Southern California School of Medicine, Los Angeles, CA 90033; and 1lnstitute de Biomedicine, Caracas 101, Venezuela Contributed by Barry R. Bloom, November 2, 1987 ABSTRACT Cell walls of Mycobacterium leprae, pre- positive healthy contacts of lepromatous patients or from pared by differential solvent extraction, were shown to contain blood or lesions of patients with tuberculoid leprosy, only a arabinogalactan, mycolates, and peptidoglycan. In addition, small proportion of clones tested are responsive to the amino acid analysis revealed the unexpected presence of large known serologically defined recombinant antigens (ref. 5; amounts of protein that retained potent immunological reac- J.M.-K., R.L.M., and B.R.B., unpublished data). tivity. Purified cell walls stimulated proliferation of T cells For many years, significant immune responses to purified -

Structures on the Cell Surface
ARTHRITIS & RHEUMATISM Volume 37 Number 8, August 1994, pp 1245-1248 0 1994, American College of Rheumatology 1245 SPECIAL ARTICLE STRUCTURES ON THE CELL SURFACE Update from the Fifth International Workshop on Human Leukocyte Differentiation Antigens NORA G. SINGER, ROBERT F. TODD, and DAVID A. FOX What is the value of the CD classification of leukocyte common surface antigens. In many instances this has surface antigens? allowed the assignment of as-yet-unclassified mono- clonal antibodies to known CD clusters. More impor- Monoclonal antibody technology has led to the identification of a large number of leukocyte surface tantly, it has permitted the definition of new surface structures over the past 15 years. More importantly, structures, for which information from any individual the antibodies themselves have proven to be powerful laboratory working with one reagent would not have tools for understanding the function and structure of been sufficient to properly characterize a new antigen. these antigens. Many of these molecules now have In some instances antibodies have been misclassified defined roles in cell signaling, adhesion, and regulation by submitting laboratories, and the workshop analyses of inflammatory processes. An in-depth understanding provide a mechanism for correctly identifying the of the function of these molecules is essential to target antigen. This potentially averts many mistakes defining their importance in inflammatory and auto- in research laboratories. It is also of value in clinical immune diseases. Hopefully, many of these structures therapeutics, since misclassified antibodies have, on a will ultimately be suitable targets for new pharmaco- rare occasion, already been in use in clinical protocols. -

Monosan Catalogus Binnenwerk.Indd
Content Antibodies for research in the following areas: Page Acrosomal Proteins 2 Cell Adhesion Molecules 3 Cell Cycle Associated Markers 15 Cell Signaling Research 17 Cluster Differentiation Markers 21 Collagens 70 Cytokines 75 Enzymes 83 Growth Factors and Receptors 86 Haematology 96 Hormones 99 Immunoglobulins 107 Keratins 110 Lymphoid Markers 116 Microbiology/Virology 124 Microtubuli 128 Multidrug Resistance Antigens 129 Muscles 135 Neuroscience 137 Special Interest 145 Tumour Markers 153 Secondary Reagents 170 Mouse Isotype Controls 171 Streptavidin 171 Aluminium Cryo-tubes for Storage in Liquid Nitrogen 172 Target Unmasking Fluid 172 General Technical Instructions for Use 173 Product Name Index 176 Product Code Index 189 Distributor Information 202 F = Frozen Sections (IHC) IP = Immunoprecipitation P = Paraffin Sections (IHC) IF = Immunofluorescence C = Flow Cytometry EM = Electron Microscopy E = ELISA A = Agglutination WB = Western Blotting B = Blocking IC = Immuno Cytology N = Neutralization Inh = Inhibition of biological activity FS = Functional Studies MC = Micro Cytotoxity Testing All antibodies are reactive with human antigens, unless stated otherwise. Acrosomal Proteins Cell Adhesion Molecules Intra-acrosomal protein, Bull Intra-acrosomal protein αvβ3 INTEGRIN β Catenin Cat.no. MON 8015 (Monoclonal) Cat.no. MON 8017 (Monoclonal) Cat.no. MON 2032 (Monoclonal) Cat. no. MON 3055 (Monoclonal) NEW Clone ACR.4 Clone Hs-14 Clone BV3 Clone 9F2 The antibody reacts with intra-acrosomal protein 28 kDa of bull The antibody reacts with human intra-acrosomal proteins (220 kDa). This monoclonal antibody (subclass IgG1) binds to human αvβ3 integrin Beta-catenin belongs together with alpha- and gamma-catenin to the spermatozoa, can be used for detection of state of acrosome of Strong reactivity was also observed with mouse sperms.