Reichel Marco.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Study on the Formation and the Decomposition of Agn3 and A
Study on the formation and the decomposition of AgN3 and a hypothetical compound ReN3 by using density functional calculations. G. Soto. Universidad Nacional Autónoma de México, Centro de Nanociencias y Nanotecnología Km 107 carretera Tijuana-Ensenada, Ensenada Baja California, México. Corresponding Author: G. Soto. CNyN-UNAM P.O. Box 439036, San Ysidro, CA 92143-9036, USA Tel: +52+646+1744602, Fax: +52+646+1744603 E-Mail: [email protected] Abstract We present a comparative study between ReN3 and AgN3 by using density functional theory. The ReN3 is a hypothetical compound proposed by us to interpret the Re to Re interplanar spacing of thin films grown by sputtering. Both, the AgN3 as the ReN3, are calculated as positive enthalpy compounds. The enthalpy might give a clue about the spontaneous decomposition of the solid form, but it cannot be interpreted as a restriction of its synthesizability. As from the calculated total-energy, we discuss the route for the formation of AgN3 starting from atomic species in aqueous solution. We propose that their synthesizability is conditioned by the energy of free nitrogen atoms, and the kinetics of reaction. We conclude that the intrinsic stability of a certain atomic arrangement depends only of the equilibrium of atomic forces, and not from the energy value associated with that structure. 1 1. Introduction Predicting new solids based solely on computer calculations is one of the main challenges of materials science. Achieving this would mean a giant step forward as it would save many hours of fruitless efforts. Although there has been significant progress[1], it is still early to sing praises. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Commerce in Explosives; 2020 Annual Those on the Annual List
Federal Register / Vol. 85, No. 247 / Wednesday, December 23, 2020 / Notices 83999 inspection at the Office of the Secretary or synonyms in brackets. This list Black powder substitutes. and on EDIS.3 supersedes the List of Explosive *Blasting agents, nitro-carbo-nitrates, This action is taken under the Materials published in the Federal including non-cap sensitive slurry and authority of section 337 of the Tariff Act Register on January 2, 2020 (Docket No. water gel explosives. of 1930, as amended (19 U.S.C. 1337), 2019R–04, 85 FR 128). Blasting caps. and of §§ 201.10 and 210.8(c) of the The 2020 List of Explosive Materials Blasting gelatin. Commission’s Rules of Practice and is a comprehensive list, but is not all- Blasting powder. Procedure (19 CFR 201.10, 210.8(c)). inclusive. The definition of ‘‘explosive BTNEC [bis (trinitroethyl) carbonate]. materials’’ includes ‘‘[e]xplosives, BTNEN [bis (trinitroethyl) nitramine]. By order of the Commission. BTTN [1,2,4 butanetriol trinitrate]. Issued: December 18, 2020. blasting agents, water gels and detonators. Explosive materials, Bulk salutes. William Bishop, include, but are not limited to, all items Butyl tetryl. Supervisory Hearings and Information in the ‘List of Explosive Materials’ Officer. C provided for in § 555.23.’’ 27 CFR Calcium nitrate explosive mixture. [FR Doc. 2020–28458 Filed 12–22–20; 8:45 am] 555.11. Accordingly, the fact that an BILLING CODE 7020–02–P Cellulose hexanitrate explosive explosive material is not on the annual mixture. list does not mean that it is not within Chlorate explosive mixtures. coverage of the law if it otherwise meets DEPARTMENT OF JUSTICE Composition A and variations. -
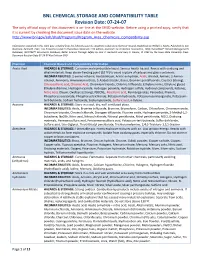
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title DETERMINATION OF AZIDE ION BY HYDROGEN ION TITRATION AFTER OXIDATION WITH NITRITE Permalink https://escholarship.org/uc/item/39q6h8sg Authors Clem, Ray G. Huffman, E.H. Publication Date 1964-11-01 eScholarship.org Powered by the California Digital Library University of California UCRL-11777 University of California Ernest 0. lawrence Radiation Laboratory DETERMINATION OF AZIDE ION BY HYDROGEN ION TITRATION AFTER OXIDATION WITH NITRITE TWO-WEEK lOAN COPY This is a library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Division, Ext. 5545 Berkeley, California DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of Califomia. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Govemment or any agency thereof or the Regents of the University of California. -

Silver and Silver Compounds: Environmental Aspects
This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organization, or the World Health Organization. Concise International Chemical Assessment Document 44 SILVER AND SILVER COMPOUNDS: ENVIRONMENTAL ASPECTS The layout and pagination of this pdf file are not necessarily identical to those of the hard copy First draft prepared by Mr P.D. Howe and Dr S. Dobson, Centre for Ecology and Hydrology, Monks Wood, United Kingdom Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organization, and the World Health Organization, and produced within the framework of the Inter-Organization Programme for the Sound Management of Chemicals. World Health Organization Geneva, 2002 The International Programme on Chemical Safety (IPCS), established in 1980, is a joint venture of the United Nations Environment Programme (UNEP), the International Labour Organization (ILO), and the World Health Organization (WHO). The overall objectives of the IPCS are to establish the scientific basis for assessment of the risk to human health and the environment from exposure to chemicals, through international peer review processes, as a prerequisite for the promotion of chemical safety, and to provide technical assistance in strengthening national capacities for the sound management of chemicals. The Inter-Organization Programme for the Sound Management of Chemicals (IOMC) was established in 1995 by UNEP, ILO, the Food and Agriculture Organization of the United Nations, WHO, the United Nations Industrial Development Organization, the United Nations Institute for Training and Research, and the Organisation for Economic Co-operation and Development (Participating Organizations), following recommendations made by the 1992 UN Conference on Environment and Development to strengthen cooperation and increase coordination in the field of chemical safety. -

Dyeing Reagents for Thin-Layer and Paper Chromatography
Spraying: Dry the chromatogram to remove the solvent, then cool. Place it vertically Dyeing Reagents for Thin-Layer and Paper into a spraying box or into a fume cupboard and protect the surroundings by covering Chromatography with filter paper or the like. Apply the spray solution from about 30 cm until the layer is evenly wetted but not for so long that the chromatogram begins to run with liquid. In most cases the chromatogram is specially treated at this stage. For details please refer to the directions quoted for the particular reagent concerned. Unless otherwise stated, Contents subsequent treatment should be taken as meaning drying at room temperature. • Introduction Dipping: In case of quantitative evaluation dipping of the chromatogram into the staining solution is becoming ever more usual with respect to precision and • Index of compounds and compound classes and appropriate detection repeatability. In general less concentrated reagent solutions prepared with less polar reagents solvents are common for dipping purposes. Care should be taken in the choice of • Reagents in alphabetical order solvent to ensure that neither the chromatographically separated substances nor their reaction products are soluble in the solvent of the dipping reagent. Introduction Ready-to-use Spray Solutions Merck sells a number of ready-to-use spray solutions in 100 ml glass bottles which can The first collection of instructions for the preparation of staining reagents was directly be connected to the rechargeable electro-pneumatically operated TLC sprayer published by K.G. Krebs, D. Heusser, and H. Wimmer in Egon Stahl’s "Handbook of (Ord. No. 1.08540): Thin-Layer Chromatography" in the sixties and lateron in a revised form repeatedly as a brochure by E. -

Chemical Hygiene Plan 03/01/10
OFFICIAL POLICY 0.000 Chemical Hygiene Plan 03/01/10 Policy Statement It is the policy of the College of Charleston teaching and research laboratories in any department that use chemicals, to comply with the letter and intent of the Occupational Safety and Health Administration’s standard for safety in laboratories. This standard (29 CFR 1910.1450), requires that employers who may expose employees to potentially hazardous chemicals in laboratories have written policies and procedures for preventing employee illness or injury due to that potential exposure. This policy satisfies this requirement and is applicable to all employees working in laboratories where such exposure is possible. _______________________________________________________________ Policy Manager and Responsible Department or Office Randy L. Beaver, Director, Environmental Health and Safety Department _______________________________________________________________ Purpose/Reason for the Policy Manage the large numbers of chemical and biological agents used in student and research laboratories at all College of Charleston facilities and locations. _______________________________________________________________ Departments/Offices Affected by the Policy Any department that uses chemicals or biological agents in the process of laboratory activity. _______________________________________________________________ Procedures Related to the Policy All procedures related to this policy are attached to this file form in a .pdf file format. _______________________________________________________________ -

Chemical Hygiene Plan
This document contains information regarding the safe handling of laboratory chemicals and was prepared to comply with OSHA standard CFR 1910.1450. It was authored by Donald Albert and Joseph Bruno, and was adapted from a similar plan in effect at Yale University; the authors acknowledge invaluable assistance rendered by Thomas C. Oimet, Yale University. Edited 9 September 2016 C. Keeler Reviewed 21 September 2016 W. Nelligan 1 Contents LIST OF EMERGENCY CALL NUMBERS ................................................................................ 6 1.0 CHEMICAL HYGIENE PLAN .......................................................................................... 7 1.1 INTRODUCTION ............................................................................................................ 7 1.2 CHEMICAL HYGIENE RESPONSIBILITIES .............................................................. 8 CHEMICAL HYGIENE HIERARCHY....................................................................................... 10 1.3 DEFINITIONS ............................................................................................................... 11 1.3.1 Laboratory Definition ............................................................................................. 11 1.3.2 Hazardous Chemical Definition .............................................................................. 11 1.4 HAZARD IDENTIFICATION ...................................................................................... 13 1.5 TRAINING & INFORMATION .................................................................................. -

Solubility Data Series
INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY ANALYTICAL CHEMISTRY DIVISION COMMISSION ON SOLUBILITY DATA SOLUBILITY DATA SERIES Volume 19 CUMULATIVE INDEX VOLUMES 1-18 SOLUBILITY DATA SERIES Volume 1 H. L. Clever, Helium and Neon Volume 2 H. L. Clever, Krypton, Xenon and Radon Volume 3 M. Salomon, Silver Azide, Cyanide, Cyanamides, Cyanate, Selenocyanate and Thiocyanqte Volume 4 H. L. Clever, Argon Volume 5/6 C. L. Young, Hydrogen and Deuterium Volume 7 R. Battino, Oxygen and Ozone Volume 8 C. L. Young, Oxides of Nitrogen Volume 9 W. Hayduk, Ethane Volume 10 R. Battino, Nitrogen and Air Volume 11 B. Scrosati and C. A. Vincent, Alkali Metal, Alkaline Earth Metal and Ammonium Halides. Amide Solvents Volume 12 C. L. Young, Sulfur Dioxide, Chlorine, Fluorine and Chlorine Oxides Volume 13 S. Siekierski, T. Mioduski and M. Salomon, Scandium, Yttrium, Lanthanum and Lanthanide Nitrates Volume 14 H. Miyamoto, M. Salomon and H. L. Clever, Alkaline Earth Metal Halates Volume 15 A. F. M. Barton, Alcohols with Water Volume 16/17 E. Tomlinson and A. Regosz, Antibiotics: I. f3-Lactam Antibiotics Volume 18 O. Popovych, Tetraphenylborates Volume 19 C. L. Young, Cumulative Index: Volumes 1-18 Selected Volumes in Preparation A. L. Horvath and F. W. Getzen, Halogenated Benzenes, Toluenes andPhenols with Water C. L. Young and P. G. T. Fogg, Ammonia, Amines, Phosphine, Arsine, Stibine, Silane, Germane andStannane in Organic Solvents T. Mioduski and M. Salomon, Scandium, Yttrium, Lanthanum andLanthanide Halides in Nonaqueous Solvents T. P. Dirkse, Copper, Silver, Gold, andZinc, Cadmium andMercury Oxides andHydroxides W. Hayduk, Propane, Butane and2-Methylpropane H. -

Energetic Materials 1
ENERGETIC MATERIALS Energetic Materials 1 Volume 1 : Physics and Chemistry of the Inorganic Azides Volume 2: Technology of the Inorganic Azides Physics and Chemistry of the Inorganic Azides Edited by H. [I. Fair :1nC1 R. F. W;~lkcr Energetic Materiuh Division Armanrent Research and Development Command Dover, New Jersey PLENUM PRESS NEW YORK AND LONDON and b + i. Some of the reactions are evidently complex, and such equations &$@[ The same author 1241 also obtained HN3 from hydrazine and hydroxylamine by oxidation (b + c): Lm text are not strictly quantitative. i 1 laterCu , discovered HN3 [I] by reacting benzoylhydrazine with nitrous acid i N2H4 + NHzOH + 20 4 HN3 + 3H2O (b + g-type reaction). The resulting benzoyl azide was saponified, and sodium azide was isolated from the alkaline mixture. In 1903 Wislicenus [14] synthesized Of various oxidants tested, hydroperoxide and chromic acid gave the best sodium azide solely from inorganic compounds, namely, sodium metal, am- results (24% yield). monia, and dinitrogen oxide (a + e). The reaction proceeds in two steps, first Thiele [25] converted aminoguanidine with nitrous acid (b + g) to guanyl converting ammonia with sodium to sodamide, and then reacting this with di- azide [26] which was saponified to sodium azide: nitrogen oxide to yield sodium azide. Fifty percent of the sodamide is decom- posed to hydroxide and ammonia, and the overall balance of the process is 2NaNH2 + N20 -+ NaN3 + NaOH + NH3 Wislicenus conducted the synthesis as a dry procedure at elevated temperature; a Another interesting reaction (b + g) was studied by Freund and Schander low-temperature procedure in liquid ammonia was later patented by Acken and [?7]. -

Guide for the Selection of Explosives Detection and Blast Mitigation Equipment for Emergency First Responders
Guide for the Selection of Explosives Detection and Blast Mitigation Equipment for Emergency First Responders Preparedness Directorate Office of Grants and Training Guide 105–07 February 2008 Homeland Security Guide for the Selection of Explosives Detection and Blast Mitigation Equipment for Emergency First Responders Guide 105–07 Dr. Alim A. Fatah1 Richard D. Arcilesi, Jr.2 Dr. Joseph A. McClintock2 Charlotte H. Lattin2 Michael Helinski2 Martin Hutchings3 Coordination by: Office of Law Enforcement Standards National Institute of Standards and Technology Gaithersburg, MD 20899–8102 Prepared for: U.S. Department of Homeland Security Preparedness Directorate Office of Grants and Training Systems Support Division 810 7th Street, NW Washington, DC 20531 February 2008 1 National Institute of Standards and Technology (NIST), Office of Law Enforcement Standards (OLES). 2 Battelle. 3 National Bomb Squad Commanders Advisory Board (NBSCAB), retired. This guide was prepared for the Preparedness Directorate’s Office of Grants and Training (G&T) Systems Support Division (SDD) by the Office of Law Enforcement Standards at the National Institute of Standards and Technology (NIST) under Interagency Agreement 94–IJ–R–004, Project No. 99–060–CBW. It was also prepared under CBIAC contract No. SP0700–00–D–3180 and Interagency Agreement M92361 between NIST and the Department of Defense Technical Information Center (DTIC). The authors wish to thank Ms. Kathleen Higgins of NIST for programmatic support and for numerous valuable discussions concerning the contents of this document. We also wish to acknowledge the InterAgency Board (IAB) for Equipment Standardization and Interoperability and the Responder Knowledge Base (RKB). The IAB (made up of government and first responder representatives) was established to ensure equipment standardization and interoperability and to oversee the research and development of advanced technologies to assist first responders at the state and local levels in establishing and maintaining a robust crisis and consequence management capability.