Neuromuscular Junctions As Key Contributors and Therapeutic Targets in Spinal Muscular Atrophy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A System for Studying Mechanisms of Neuromuscular Junction Development and Maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R
© 2016. Published by The Company of Biologists Ltd | Development (2016) 143, 2464-2477 doi:10.1242/dev.130278 TECHNIQUES AND RESOURCES RESEARCH ARTICLE A system for studying mechanisms of neuromuscular junction development and maintenance Valérie Vilmont1,‡, Bruno Cadot1, Gilles Ouanounou2 and Edgar R. Gomes1,3,*,‡ ABSTRACT different animal models and cell lines (Chen et al., 2014; Corti et al., The neuromuscular junction (NMJ), a cellular synapse between a 2012; Lenzi et al., 2015) with the hope of recapitulating some motor neuron and a skeletal muscle fiber, enables the translation of features of neuromuscular diseases and understanding the triggers chemical cues into physical activity. The development of this special of one of their common hallmarks: the disruption of the structure has been subject to numerous investigations, but its neuromuscular junction (NMJ). The NMJ is one of the most complexity renders in vivo studies particularly difficult to perform. studied synapses. It is formed of three key elements: the lower motor In vitro modeling of the neuromuscular junction represents a powerful neuron (the pre-synaptic compartment), the skeletal muscle (the tool to delineate fully the fine tuning of events that lead to subcellular post-synaptic compartment) and the Schwann cell (Sanes and specialization at the pre-synaptic and post-synaptic sites. Here, we Lichtman, 1999). The NMJ is formed in a step-wise manner describe a novel heterologous co-culture in vitro method using rat following a series of cues involving these three cellular components spinal cord explants with dorsal root ganglia and murine primary and its role is basically to ensure the skeletal muscle functionality. -

The Histology of the Neuromuscular Junction In
75 THE HISTOLOGY OF THE NEUROMUSCULAR JUNCTION Downloaded from https://academic.oup.com/brain/article/84/1/75/372729 by guest on 27 September 2021 IN DYSTROPHIA MYOTONICA BY VIOLET MACDERMOT Department of Neurology, St. Thomas' Hospital, London, S.E.I (1) INTRODUCTION DYSTROPJHC MYOTONICA is a familial disease affecting males and females, usually presenting in adult life, characterized by muscular wasting and weakness together with certain other features. The muscles mainly in- volved are the temporal, masseter, facial, sternomastoid and limb muscles, in the latter those mainly affected being peripheral in distribution. A widespread disorder of muscular contraction, myotonia, is also present but is noticed chiefly in the tongue and in the muscles involved in grasping. The other features of the condition are some degree of mental defect, dysphonia, cataracts, frontal baldness, sparse body hair and testicular atrophy. Any of the manifestations of the disease may be absent and the order of presentation of symptoms is variable. The myotonia may precede muscular wasting by many years or may occur independently. In those muscles which are severely wasted the myotonia tends to disappear. The interest of dystrophia myotonica lies in the peculiar distribution of muscle involvement and in the combination of a disorder of muscle function with endocrine and other dysplasic features. The results of histological examination of biopsy and post-mortem material have been described and reviewed by numerous workers, notably Steinert (1909), Adie and Greenfield (1923), Keschner and Davison (1933), Hassin and Kesert (1948), Wohlfart (1951), Adams, Denny-Brown and Pearson (1953), Greenfield, Shy, Alvord and Berg (1957). -
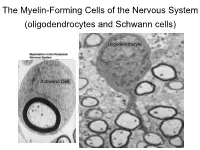
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Specific Labeling of Synaptic Schwann Cells Reveals Unique Cellular And
RESEARCH ARTICLE Specific labeling of synaptic schwann cells reveals unique cellular and molecular features Ryan Castro1,2,3, Thomas Taetzsch1,2, Sydney K Vaughan1,2, Kerilyn Godbe4, John Chappell4, Robert E Settlage5, Gregorio Valdez1,2,6* 1Department of Molecular Biology, Cellular Biology, and Biochemistry, Brown University, Providence, United States; 2Center for Translational Neuroscience, Robert J. and Nancy D. Carney Institute for Brain Science and Brown Institute for Translational Science, Brown University, Providence, United States; 3Neuroscience Graduate Program, Brown University, Providence, United States; 4Fralin Biomedical Research Institute at Virginia Tech Carilion, Roanoke, United States; 5Department of Advanced Research Computing, Virginia Tech, Blacksburg, United States; 6Department of Neurology, Warren Alpert Medical School of Brown University, Providence, United States Abstract Perisynaptic Schwann cells (PSCs) are specialized, non-myelinating, synaptic glia of the neuromuscular junction (NMJ), that participate in synapse development, function, maintenance, and repair. The study of PSCs has relied on an anatomy-based approach, as the identities of cell-specific PSC molecular markers have remained elusive. This limited approach has precluded our ability to isolate and genetically manipulate PSCs in a cell specific manner. We have identified neuron-glia antigen 2 (NG2) as a unique molecular marker of S100b+ PSCs in skeletal muscle. NG2 is expressed in Schwann cells already associated with the NMJ, indicating that it is a marker of differentiated PSCs. Using a newly generated transgenic mouse in which PSCs are specifically labeled, we show that PSCs have a unique molecular signature that includes genes known to play critical roles in *For correspondence: PSCs and synapses. These findings will serve as a springboard for revealing drivers of PSC [email protected] differentiation and function. -

Neuromuscular Junctions LEARNING OBJECTIVES: ➢ Components of the Neuromuscular Junction (NMJ) ➢ Physiological Anatomy of NMJ
NEUROMUSCULAR JUNCTION By:Dr.Fareeda banu A.B. Associate professor Dept of Physiology, USM-KLE IMP Neuromuscular Junctions LEARNING OBJECTIVES: ➢ Components of the neuromuscular junction (NMJ) ➢ Physiological anatomy of NMJ. ➢ Synthesis, storage and release of Ach at NMJ ➢ Events occurring during neuromuscular transmission with reference to end-plate potentials. ➢ Clinical importance of NMJ ➢ Drugs affecting NMJ transmission. ➢ Applied aspects. INTRODUCTION Movement of the body requires the interaction of a complex series of afferent and efferent neuronal signals, which result in a skeletal muscle contraction The neuromuscular junction is a specialized form of a chemical synapse comprised of an alpha motor neuron and the muscle fiber it innervates. INTRODUCTION Junctions Vrs. Synapses - NMJ is a junction between a motor neuron and a skeletal muscle. Synapse is a connection between two neurons A junction will always respond to an action potential in the presynaptic nerve. Synapses may or may not. A junction will have a safety factor, sufficient release of neurotransmitter to insure action potential generation in the effector organ, usually a thousand or so times over. MOTOR UNIT: THE NERVE-MUSCLE FUNCTIONAL UNIT ❖ Motor Unit: The Nerve-Muscle Functional Unit. A motor unit is a motor neuron and all the muscle fibers it innervate ❖ The nerve fiber forms a complex of branching nerve terminals that invaginate into the surface of the muscle fiber but lie outside the muscle fiber plasma membrane. The entire structure is called the motor end plate. NEUROMUSCULAR JUNCTION (NMJ) Defn: NMJ is a junction between the motor nerve ending and skeletal muscle fiber. Components of the NMJ: It is comprised of; Unmyelinated terminal boutons of axon supplying a skeletal muscle fiber. -

Chapter 7 Excitation of Skeletal Muscle: Neuromuscular Transmission and Excitation-Contraction Coupling
C H A P T E R 7 U N I T I I Excitation of Skeletal Muscle: Neuromuscular Transmission and Excitation-Contraction Coupling TRANSMISSION OF IMPULSES cytoplasm of the terminal, but it is absorbed rapidly into FROM NERVE ENDINGS TO many small synaptic vesicles, about 300,000 of which are SKELETAL MUSCLE FIBERS: THE normally in the terminals of a single end plate. In the syn- NEUROMUSCULAR JUNCTION aptic space are large quantities of the enzyme acetylcho- linesterase, which destroys acetylcholine a few milliseconds Skeletal muscle fibers are innervated by large, myelinated after it has been released from the synaptic vesicles. nerve fibers that originate from large motoneurons in the anterior horns of the spinal cord. As discussed in Chapter SECRETION OF ACETYLCHOLINE 6, each nerve fiber, after entering the muscle belly, nor- BY THE NERVE TERMINALS mally branches and stimulates from three to several hundred skeletal muscle fibers. Each nerve ending makes When a nerve impulse reaches the neuromuscular junc- a junction, called the neuromuscular junction, with the tion, about 125 vesicles of acetylcholine are released from muscle fiber near its midpoint. The action potential initi- the terminals into the synaptic space. Some of the details ated in the muscle fiber by the nerve signal travels in both of this mechanism can be seen in Figure 7-2, which directions toward the muscle fiber ends. With the excep- shows an expanded view of a synaptic space with the tion of about 2 percent of the muscle fibers, there is only neural membrane above and the muscle membrane and one such junction per muscle fiber. -

A Mathematical Model of the Neuromuscular Junction and Muscle Force Generation in the Pathological Condition Myasthenia Gravis
A Mathematical Model of the Neuromuscular Junction and Muscle Force Generation in the Pathological Condition Myasthenia Gravis Taylor Meredith Abstract At the neuromuscular junction, motor neurons stimulate muscle fibers to contract through many detailed processes. This paper discusses a model to connect the many processes involved in muscle contraction from the electrical activity of nerve impulses to the mechanical force generation. Through the use of differential equations to describe calcium dynamics and muscle force, coupled to end plate potentials at the neuromuscular junction, we model this process in both a healthy system and a damaged system. As an application of our model, we focus on the neuromuscular disease myasthenia gravis and how it affects muscle force generation in our model, as well as the effect of acetylcholinesterase inhibiting treatment. 1 Introduction Figure 1: The neuromuscular junction. Our goal is to design a mathematical model that bridges the gap between the electrical activity of neurons and the mechanical action of muscle contraction. This so-called \gap" is known as the neuromuscular junction (Figure 1), an important biological feature in humans, as well as other animals, that allows for the transmission of signals from a motor neuron to a muscle fiber [7]. The motor neuron axon is a long projection of the motor neuron that conducts electrical 1 impulses known as action potentials. At every muscle fiber a neuromuscular junction exists, where the motor neuron axon releases synaptic vesicles, each containing a `quantum' of the neurotransmitter. In mammals, the neurotransmitter is acetylcholine and each `quantum' contains approximately 10,000 molecules of acetylcholine [6]. -

The Neuromuscular Junction: Roles in Aging and Neuromuscular Disease
International Journal of Molecular Sciences Review The Neuromuscular Junction: Roles in Aging and Neuromuscular Disease Shama R. Iyer 1, Sameer B. Shah 2 and Richard M. Lovering 1,* 1 Department of Orthopaedics, University of Maryland School of Medicine, AHB, Room 540, 100 Penn St., Baltimore, MD 21201, USA; [email protected] 2 Departments of Orthopaedic Surgery and Bioengineering, University of California San Diego, La Jolla, CA 92093, USA; [email protected] * Correspondence: [email protected] Abstract: The neuromuscular junction (NMJ) is a specialized synapse that bridges the motor neuron and the skeletal muscle fiber and is crucial for conversion of electrical impulses originating in the mo- tor neuron to action potentials in the muscle fiber. The consideration of contributing factors to skeletal muscle injury, muscular dystrophy and sarcopenia cannot be restricted only to processes intrinsic to the muscle, as data show that these conditions incur denervation-like findings, such as fragmented NMJ morphology and corresponding functional changes in neuromuscular transmission. Primary defects in the NMJ also influence functional loss in motor neuron disease, congenital myasthenic syndromes and myasthenia gravis, resulting in skeletal muscle weakness and heightened fatigue. Such findings underscore the role that the NMJ plays in neuromuscular performance. Regardless of cause or effect, functional denervation is now an accepted consequence of sarcopenia and muscle disease. In this short review, we provide an overview of the pathologic etiology, symptoms, and Citation: Iyer, S.R.; Shah, S.B.; therapeutic strategies related to the NMJ. In particular, we examine the role of the NMJ as a disease Lovering, R.M. -

The Somatic Nervous System Mimi Jakoi, Phd Jennifer Carbrey, Phd
Introductory Human Physiology ©copyright Jennifer Carbrey & Emma Jakoi The Somatic Nervous System Mimi Jakoi, PhD Jennifer Carbrey, PhD The underlined headings correspond to the two Somatic Nervous system videos. 1. Introduction and structure The efferent portion of the peripheral nervous system consists of the somatic nervous system and the autonomic nervous system. The autonomic nervous system controls the function of glands, smooth muscle, cardiac muscle, and the neurons of the GI tract. It is composed of two neurons in series that can either excite or inhibit the target organ. In contrast, the somatic nervous system contains single neurons that excite skeletal muscles. The movements controlled by the somatic nervous system can be voluntary or involuntary (reflexes). Motor Unit The axons of motor neurons are myelinated and have large diameters for fast conduction of action potentials. As the axon approaches a skeletal muscle fiber (muscle cell) it usually branches to form synapses with anywhere from three to one thousand muscle fibers. However, each muscle fiber is usually innervated by only a single neuron. A motor unit consists of a neuron and all of the muscle fibers it innervates. A single neuron innervates fibers from only one muscle and the innervated muscle fibers are usually spread throughout the muscle. The portion of the skeletal muscle fiber plasma membrane that synapses with the motor neuron axon is called the motor end plate. Once an action potential arrives at the axon terminal, the depolarization of the membrane opens voltage-gated calcium channels (Fig. 1). An increase in intracellular calcium at the terminal causes release of acetylcholine vesicles into the neuromuscular junction. -

Loss of Glialneurofascin155delays Developmental Synapse
12904 • The Journal of Neuroscience, September 17, 2014 • 34(38):12904–12918 Development/Plasticity/Repair Loss of Glial Neurofascin155 Delays Developmental Synapse Elimination at the Neuromuscular Junction Sarah L. Roche,1,2 Diane L. Sherman,3 Kosala Dissanayake,1,4 Genevie`ve Soucy,5 Anne Desmazieres,3 X Douglas J. Lamont,6 Elior Peles,7 Jean-Pierre Julien,5 Thomas M. Wishart,2,8 Richard R. Ribchester,1,2 Peter J. Brophy,3* and Thomas H. Gillingwater1,2* 1Centre for Integrative Physiology, University of Edinburgh, Edinburgh, EH8 9XF, United Kingdom, 2Euan MacDonald Centre for Motor Neuron Disease Research, University of Edinburgh, Edinburgh, EH16 4SB, United Kingdom, 3Centre for Neuroregeneration, University of Edinburgh, Edinburgh, EH16 4SB, United Kingdom, 4Clinical Pharmacology Unit, University of Edinburgh, Queen’s Medical Research Institute, Edinburgh, EH16 4TJ, United Kingdom, 5Research Centre of Centre Hospitalier Universitaire de Que´bec, Department of Psychiatry and Neurosciences, Laval University, Que´bec F-3471, Canada, 6FingerPrints Proteomics Facility, College of Life Sciences, University of Dundee, Dundee, DD1 5EH, United Kingdom, 7Department of Molecular Cell Biology, Weizmann Institute of Science, POB 26, Rehovot 76100, Israel, and 8Division of Neurobiology, Roslin Institute and Royal Dick School of Veterinary Studies, University of Edinburgh, Edinburgh, EH25 9RG, United Kingdom Postnatal synapse elimination plays a critical role in sculpting and refining neural connectivity throughout the central and peripheral nervous systems, including the removal of supernumerary axonal inputs from neuromuscular junctions (NMJs). Here, we reveal a novel and important role for myelinating glia in regulating synapse elimination at the mouse NMJ, where loss of a single glial cell protein, the glial isoform of neurofascin (Nfasc155), was sufficient to disrupt postnatal remodeling of synaptic circuitry. -

Regulation of Motor Axon Innervation at the Neuromuscular Junction By
Regulation of Motor Axon Innervation at the Neuromuscular Junction by Jennifer L. Shadrach A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Cellular and Molecular Biology) in the University of Michigan 2017 Doctoral Committee: Associate Professor Brian A. Pierchala, Chair Associate Professor Anthony Antonellis Professor Susan V. Brooks Professor Roman J. Giger Professor Daniel Goldman Jennifer L. Shadrach [email protected] ORCID iD: 0000-0001-5266-8625 © Jennifer L. Shadrach 2017 ACKNOWLEDGEMENTS: First, I would like to thank Dr. Brian Pierchala for the opportunity to work in your laboratory. I have benefited greatly from your guidance and support over the past five years. Additionally, I appreciate your willingness in allowing me to explore my scientific interests and develop into an independent researcher. What I have learned from my experiences will be extremely valuable as I pursue my future scientific career. I would also like to thank the entire Pierchala lab for their help and insights over the years. In particular, I would like thank Allison Milan for her technical assistance throughout this past year. Finally, a special thanks to Chris Donnelly – without both your scientific input and friendship over the years my graduate school experience would have not been the same. To my committee members, thank you for your time, wisdom, and guidance throughout my graduate training. Your thoughtful comments and advice, as well as your willingness to share various protocols and reagents have been greatly appreciated. Finally, I would like to thank the Cellular and Molecular Biology Program and all my friends and colleagues for supporting me throughout graduate school career.