U5 Snrna Interactions with Exons Ensure Splicing Precision 2 3 Olga V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
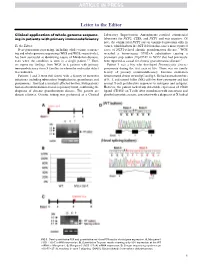
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases
G C A T T A C G G C A T genes Review RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases Amber Willbanks, Shaun Wood and Jason X. Cheng * Department of Pathology, Hematopathology Section, University of Chicago, Chicago, IL 60637, USA; [email protected] (A.W.); [email protected] (S.W.) * Correspondence: [email protected] Abstract: Chromatin structure plays an essential role in eukaryotic gene expression and cell identity. Traditionally, DNA and histone modifications have been the focus of chromatin regulation; however, recent molecular and imaging studies have revealed an intimate connection between RNA epigenetics and chromatin structure. Accumulating evidence suggests that RNA serves as the interplay between chromatin and the transcription and splicing machineries within the cell. Additionally, epigenetic modifications of nascent RNAs fine-tune these interactions to regulate gene expression at the co- and post-transcriptional levels in normal cell development and human diseases. This review will provide an overview of recent advances in the emerging field of RNA epigenetics, specifically the role of RNA modifications and RNA modifying proteins in chromatin remodeling, transcription activation and RNA processing, as well as translational implications in human diseases. Keywords: 5’ cap (5’ cap); 7-methylguanosine (m7G); R-loops; N6-methyladenosine (m6A); RNA editing; A-to-I; C-to-U; 2’-O-methylation (Nm); 5-methylcytosine (m5C); NOL1/NOP2/sun domain Citation: Willbanks, A.; Wood, S.; (NSUN); MYC Cheng, J.X. RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases. Genes 2021, 12, 627. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Growth and Gene Expression Profile Analyses of Endometrial Cancer Cells Expressing Exogenous PTEN
[CANCER RESEARCH 61, 3741–3749, May 1, 2001] Growth and Gene Expression Profile Analyses of Endometrial Cancer Cells Expressing Exogenous PTEN Mieko Matsushima-Nishiu, Motoko Unoki, Kenji Ono, Tatsuhiko Tsunoda, Takeo Minaguchi, Hiroyuki Kuramoto, Masato Nishida, Toyomi Satoh, Toshihiro Tanaka, and Yusuke Nakamura1 Laboratories of Molecular Medicine [M. M-N., M. U., K. O., T. M., T. Ta., Y. N.] and Genome Database [T. Ts.], Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan; Department of Obstetrics and Gynecology, School of Medicine, Kitasato University, Sagamihara 228-8555, Japan [H. K.]; Department of Obstetrics and Gynecology, Institute of Clinical Medicine, University of Tsukuba, Tsukuba 305-8576, Japan [M. N.]; and Department of Obstetrics and Gynecology, Ibaraki Seinan Central Hospital, Tsukuba 306-0433, Japan [T. S.] ABSTRACT Akt/protein kinase B, cell survival, and cell proliferation (8). Over- expression of PTEN can decrease cell proliferation and tumorigenicity The PTEN tumor suppressor gene encodes a multifunctional phospha- (9, 10), an observation attributed to the ability of PTEN to induce cell tase that plays an important role in inhibiting the phosphatidylinositol-3- cycle arrest and apoptosis (11, 12). kinase pathway and downstream functions that include activation of Akt/protein kinase B, cell survival, and cell proliferation. Enforced ex- Thus, lack of PTEN expression may affect a complex set of pression of PTEN in various cancer cell lines decreases cell proliferation transcriptional targets. However, no systematic assessment of PTEN- through arrest of the cell cycle, accompanied in some cases by induction regulated targets in cancer cells has been reported to date. -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

(Bsc Zoology and Microbiology) Concept of Introns and Exons
Unit-5 Molecular Biology (BSc Zoology and Microbiology) Concept of introns and exons Most of the portion of a gene in higher eukaryotes consists of noncoding DNA that interrupts the relatively short segments of coding DNA. The coding sequences are called exons. The noncoding sequences are called introns. Intron: An intron is a portion of a gene that does not code for amino acids An intron is any nucleotide sequence within a gene which is represented in the primary transcript of the gene, but not present in the final processed form. In other words, Introns are noncoding regions of an RNA transcript which are eliminated by splicing before translation. Sequences that are joined together in the final mature RNA after RNA splicing are exons. Introns are very large chunks of RNA within a messenger RNA molecule that interfere with the code of the exons. And these introns get removed from the RNA molecule to leave a string of exons attached to each other so that the appropriate amino acids can be encoded for. Introns are rare in genes of prokaryotes. #Look carefully at the diagram above, we have already discussed about the modification and processing of eukaryotic RNA. In which 5’ guanine cap and 3’poly A tail is added. So at that time, noncoding regions i.e. introns are removed. We hv done ths already. Ok Exon: The coding sequences are called Exon. An exon is the portion of a gene that codes for amino acids. In the cells of plants and animals, most gene sequences are broken up by one or more DNA sequences called introns. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Identification of Molecular Targets in Head and Neck Squamous Cell Carcinomas Based on Genome-Wide Gene Expression Profiling
1489-1497 7/11/07 18:41 Page 1489 ONCOLOGY REPORTS 18: 1489-1497, 2007 Identification of molecular targets in head and neck squamous cell carcinomas based on genome-wide gene expression profiling SATOYA SHIMIZU1,2, NAOHIKO SEKI2, TAKASHI SUGIMOTO2, SHIGETOSHI HORIGUCHI1, HIDEKI TANZAWA3, TOYOYUKI HANAZAWA1 and YOSHITAKA OKAMOTO1 Departments of 1Otorhinolaryngology, 2Functional Genomics and 3Clinical Molecular Biology, Graduate School of Medicine, Chiba University, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received May 21, 2007; Accepted June 28, 2007 Abstract. DNA amplifications activate oncogenes and are patients and metastases develop in 15-25% of patients (1). hallmarks of nearly all advanced cancers including head and Many factors, such as TNM stage, pathological grade and neck squamous cell carcinoma (HNSCC). Some oncogenes tumor site, influence the prognosis of HNSCC but are not show both DNA copy number gain and mRNA overexpression. sufficient to predict outcome. In addition, treatment often Chromosomal comparative genomic hybridization and oligo- results in impairment of functions such as speech and nucleotide microarrays were used to examine 8 HNSCC cell swallowing, cosmetic disfiguration and mental pain. These lines and a plot of gene expression levels relative to their inflictions significantly erode quality of life. To overcome this position on the chromosome was produced. Three highly situation, there is a need to find novel biomarkers that classify up-regulated genes, NT5C3, ANLN and INHBA, were patients into prognostic groups, to aid identification of high- identified on chromosome 7p14. These genes were subjected risk patients who may benefit from different treatments. to quantitative real-time RT-PCR on cDNA and genomic Comparative genomic hybridization (CGH) has facilitated DNA derived from 8 HNSCC cell lines.