Interfacing Q-Chem and CHARMM to Perform QM/MM Reaction Path Calculations*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MODELLER 10.1 Manual
MODELLER A Program for Protein Structure Modeling Release 10.1, r12156 Andrej Saliˇ with help from Ben Webb, M.S. Madhusudhan, Min-Yi Shen, Guangqiang Dong, Marc A. Martı-Renom, Narayanan Eswar, Frank Alber, Maya Topf, Baldomero Oliva, Andr´as Fiser, Roberto S´anchez, Bozidar Yerkovich, Azat Badretdinov, Francisco Melo, John P. Overington, and Eric Feyfant email: modeller-care AT salilab.org URL https://salilab.org/modeller/ 2021/03/12 ii Contents Copyright notice xxi Acknowledgments xxv 1 Introduction 1 1.1 What is Modeller?............................................. 1 1.2 Modeller bibliography....................................... .... 2 1.3 Obtainingandinstallingtheprogram. .................... 3 1.4 Bugreports...................................... ............ 4 1.5 Method for comparative protein structure modeling by Modeller ................... 5 1.6 Using Modeller forcomparativemodeling. ... 8 1.6.1 Preparinginputfiles . ............. 8 1.6.2 Running Modeller ......................................... 9 2 Automated comparative modeling with AutoModel 11 2.1 Simpleusage ..................................... ............ 11 2.2 Moreadvancedusage............................... .............. 12 2.2.1 Including water molecules, HETATM residues, and hydrogenatoms .............. 12 2.2.2 Changing the default optimization and refinement protocol ................... 14 2.2.3 Getting a very fast and approximate model . ................. 14 2.2.4 Building a model from multiple templates . .................. 15 2.2.5 Buildinganallhydrogenmodel -

Supporting Information
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2020 Supporting Information How to Select Ionic Liquids as Extracting Agent Systematically? Special Case Study for Extractive Denitrification Process Shurong Gaoa,b,c,*, Jiaxin Jina,b, Masroor Abroc, Ruozhen Songc, Miao Hed, Xiaochun Chenc,* a State Key Laboratory of Alternate Electrical Power System with Renewable Energy Sources, North China Electric Power University, Beijing, 102206, China b Research Center of Engineering Thermophysics, North China Electric Power University, Beijing, 102206, China c Beijing Key Laboratory of Membrane Science and Technology & College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, PR China d Office of Laboratory Safety Administration, Beijing University of Technology, Beijing 100124, China * Corresponding author, Tel./Fax: +86-10-6443-3570, E-mail: [email protected], [email protected] 1 COSMO-RS Computation COSMOtherm allows for simple and efficient processing of large numbers of compounds, i.e., a database of molecular COSMO files; e.g. the COSMObase database. COSMObase is a database of molecular COSMO files available from COSMOlogic GmbH & Co KG. Currently COSMObase consists of over 2000 compounds including a large number of industrial solvents plus a wide variety of common organic compounds. All compounds in COSMObase are indexed by their Chemical Abstracts / Registry Number (CAS/RN), by a trivial name and additionally by their sum formula and molecular weight, allowing a simple identification of the compounds. We obtained the anions and cations of different ILs and the molecular structure of typical N-compounds directly from the COSMObase database in this manuscript. -

GROMACS: Fast, Flexible, and Free
GROMACS: Fast, Flexible, and Free DAVID VAN DER SPOEL,1 ERIK LINDAHL,2 BERK HESS,3 GERRIT GROENHOF,4 ALAN E. MARK,4 HERMAN J. C. BERENDSEN4 1Department of Cell and Molecular Biology, Uppsala University, Husargatan 3, Box 596, S-75124 Uppsala, Sweden 2Stockholm Bioinformatics Center, SCFAB, Stockholm University, SE-10691 Stockholm, Sweden 3Max-Planck Institut fu¨r Polymerforschung, Ackermannweg 10, D-55128 Mainz, Germany 4Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, NL-9747 AG Groningen, The Netherlands Received 12 February 2005; Accepted 18 March 2005 DOI 10.1002/jcc.20291 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: This article describes the software suite GROMACS (Groningen MAchine for Chemical Simulation) that was developed at the University of Groningen, The Netherlands, in the early 1990s. The software, written in ANSI C, originates from a parallel hardware project, and is well suited for parallelization on processor clusters. By careful optimization of neighbor searching and of inner loop performance, GROMACS is a very fast program for molecular dynamics simulation. It does not have a force field of its own, but is compatible with GROMOS, OPLS, AMBER, and ENCAD force fields. In addition, it can handle polarizable shell models and flexible constraints. The program is versatile, as force routines can be added by the user, tabulated functions can be specified, and analyses can be easily customized. Nonequilibrium dynamics and free energy determinations are incorporated. Interfaces with popular quantum-chemical packages (MOPAC, GAMES-UK, GAUSSIAN) are provided to perform mixed MM/QM simula- tions. The package includes about 100 utility and analysis programs. -

Chem Compute Quickstart
Chem Compute Quickstart Chem Compute is maintained by Mark Perri at Sonoma State University and hosted on Jetstream at Indiana University. The Chem Compute URL is https://chemcompute.org/. We will use Chem Compute as the frontend for running electronic structure calculations with The General Atomic and Molecular Electronic Structure System, GAMESS (http://www.msg.ameslab.gov/gamess/). Chem Compute also provides access to other computational chemistry resources including PSI4 and the molecular dynamics packages TINKER and NAMD, though we will not be using those resource at this time. Follow this link, https://chemcompute.org/gamess/submit, to directly access the Chem Compute GAMESS guided submission interface. If you are a returning Chem Computer user, please log in now. If your University is part of the InCommon Federation you can log in without registering by clicking Login then "Log In with Google or your University" – select your University from the dropdown list. Otherwise if this is your first time working with Chem Compute, please register as a Chem Compute user by clicking the “Register” link in the top-right corner of the page. This gives you access to all of the computational resources available at ChemCompute.org and will allow you to maintain copies of your calculations in your user “Dashboard” that you can refer to later. Registering also helps track usage and obtain the resources needed to continue providing its service. When logged in with the GAMESS-Submit tabs selected, an instruction section appears on the left side of the page with instructions for several different kinds of calculations. -

Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development
processes Review Molecular Dynamics Simulations in Drug Discovery and Pharmaceutical Development Outi M. H. Salo-Ahen 1,2,* , Ida Alanko 1,2, Rajendra Bhadane 1,2 , Alexandre M. J. J. Bonvin 3,* , Rodrigo Vargas Honorato 3, Shakhawath Hossain 4 , André H. Juffer 5 , Aleksei Kabedev 4, Maija Lahtela-Kakkonen 6, Anders Støttrup Larsen 7, Eveline Lescrinier 8 , Parthiban Marimuthu 1,2 , Muhammad Usman Mirza 8 , Ghulam Mustafa 9, Ariane Nunes-Alves 10,11,* , Tatu Pantsar 6,12, Atefeh Saadabadi 1,2 , Kalaimathy Singaravelu 13 and Michiel Vanmeert 8 1 Pharmaceutical Sciences Laboratory (Pharmacy), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland; ida.alanko@abo.fi (I.A.); rajendra.bhadane@abo.fi (R.B.); parthiban.marimuthu@abo.fi (P.M.); atefeh.saadabadi@abo.fi (A.S.) 2 Structural Bioinformatics Laboratory (Biochemistry), Åbo Akademi University, Tykistökatu 6 A, Biocity, FI-20520 Turku, Finland 3 Faculty of Science-Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, 3584 CH Utrecht, The Netherlands; [email protected] 4 Swedish Drug Delivery Forum (SDDF), Department of Pharmacy, Uppsala Biomedical Center, Uppsala University, 751 23 Uppsala, Sweden; [email protected] (S.H.); [email protected] (A.K.) 5 Biocenter Oulu & Faculty of Biochemistry and Molecular Medicine, University of Oulu, Aapistie 7 A, FI-90014 Oulu, Finland; andre.juffer@oulu.fi 6 School of Pharmacy, University of Eastern Finland, FI-70210 Kuopio, Finland; maija.lahtela-kakkonen@uef.fi (M.L.-K.); tatu.pantsar@uef.fi -

Cygwin User's Guide
Cygwin User’s Guide Cygwin User’s Guide ii Copyright © Cygwin authors Permission is granted to make and distribute verbatim copies of this documentation provided the copyright notice and this per- mission notice are preserved on all copies. Permission is granted to copy and distribute modified versions of this documentation under the conditions for verbatim copying, provided that the entire resulting derived work is distributed under the terms of a permission notice identical to this one. Permission is granted to copy and distribute translations of this documentation into another language, under the above conditions for modified versions, except that this permission notice may be stated in a translation approved by the Free Software Foundation. Cygwin User’s Guide iii Contents 1 Cygwin Overview 1 1.1 What is it? . .1 1.2 Quick Start Guide for those more experienced with Windows . .1 1.3 Quick Start Guide for those more experienced with UNIX . .1 1.4 Are the Cygwin tools free software? . .2 1.5 A brief history of the Cygwin project . .2 1.6 Highlights of Cygwin Functionality . .3 1.6.1 Introduction . .3 1.6.2 Permissions and Security . .3 1.6.3 File Access . .3 1.6.4 Text Mode vs. Binary Mode . .4 1.6.5 ANSI C Library . .4 1.6.6 Process Creation . .5 1.6.6.1 Problems with process creation . .5 1.6.7 Signals . .6 1.6.8 Sockets . .6 1.6.9 Select . .7 1.7 What’s new and what changed in Cygwin . .7 1.7.1 What’s new and what changed in 3.2 . -

The Focus - Issue 36
Contents The Focus - Issue 36 A Publication for ANSYS Users Contents Feature Articles ● Linux & ANSYS: Lessons Learned ● Backup Tool ● Design Modeler FAQ On the Web ● APDL Customization course notes now available for purchase ● ANSYS and MathCAD ● ANSYS Acquires Century Dynamics Resources ● PADT Support: How can we help? ● Upcoming Training at PADT ● About The Focus ❍ The Focus Library ❍ Contributor Information ❍ Subscribe / Unsubscribe ❍ Legal Disclaimer http://www.padtinc.com/epubs/focus/common/contents.asp [3/28/2005 9:06:12 AM] Linux & ANSYS: Lessons Learned The Focus - Issue 36 A Publication for ANSYS Users Linux & ANSYS: Lessons Learned by Eric Miller, PADT Every couple of years, the computing picture for analysts gets turned upside down. For a long time now the industry has been moving from Unix workstations to Windows/Intel desktop machines. The wintel price/performance has been fantastic, the IT guys are happier, and all of that productivity software that you spend so much time with runs in the same spot. We have been happy with a stable and known environment. However, accepting the fact that unless you work for a big company that can buy some Unix servers, you just don’t have an easy way to get some extra horsepower other then getting a new box. Then along comes this Finnish guy that may or may not have been named after Lucy’s little brother. With not much of a life and a very large brain, he popped out the majority of a complete and free version of Unix that anyone can use, breaking the stranglehold of (expensive) proprietary Unix OS’s that ran on (expensive) proprietary hardware. -

Starting SCF Calculations by Superposition of Atomic Densities
Starting SCF Calculations by Superposition of Atomic Densities J. H. VAN LENTHE,1 R. ZWAANS,1 H. J. J. VAN DAM,2 M. F. GUEST2 1Theoretical Chemistry Group (Associated with the Department of Organic Chemistry and Catalysis), Debye Institute, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands 2CCLRC Daresbury Laboratory, Daresbury WA4 4AD, United Kingdom Received 5 July 2005; Accepted 20 December 2005 DOI 10.1002/jcc.20393 Published online in Wiley InterScience (www.interscience.wiley.com). Abstract: We describe the procedure to start an SCF calculation of the general type from a sum of atomic electron densities, as implemented in GAMESS-UK. Although the procedure is well known for closed-shell calculations and was already suggested when the Direct SCF procedure was proposed, the general procedure is less obvious. For instance, there is no need to converge the corresponding closed-shell Hartree–Fock calculation when dealing with an open-shell species. We describe the various choices and illustrate them with test calculations, showing that the procedure is easier, and on average better, than starting from a converged minimal basis calculation and much better than using a bare nucleus Hamiltonian. © 2006 Wiley Periodicals, Inc. J Comput Chem 27: 926–932, 2006 Key words: SCF calculations; atomic densities Introduction hrstuhl fur Theoretische Chemie, University of Kahrlsruhe, Tur- bomole; http://www.chem-bio.uni-karlsruhe.de/TheoChem/turbo- Any quantum chemical calculation requires properly defined one- mole/),12 GAMESS(US) (Gordon Research Group, GAMESS, electron orbitals. These orbitals are in general determined through http://www.msg.ameslab.gov/GAMESS/GAMESS.html, 2005),13 an iterative Hartree–Fock (HF) or Density Functional (DFT) pro- Spartan (Wavefunction Inc., SPARTAN: http://www.wavefun. -

Parameterizing a Novel Residue
University of Illinois at Urbana-Champaign Luthey-Schulten Group, Department of Chemistry Theoretical and Computational Biophysics Group Computational Biophysics Workshop Parameterizing a Novel Residue Rommie Amaro Brijeet Dhaliwal Zaida Luthey-Schulten Current Editors: Christopher Mayne Po-Chao Wen February 2012 CONTENTS 2 Contents 1 Biological Background and Chemical Mechanism 4 2 HisH System Setup 7 3 Testing out your new residue 9 4 The CHARMM Force Field 12 5 Developing Topology and Parameter Files 13 5.1 An Introduction to a CHARMM Topology File . 13 5.2 An Introduction to a CHARMM Parameter File . 16 5.3 Assigning Initial Values for Unknown Parameters . 18 5.4 A Closer Look at Dihedral Parameters . 18 6 Parameter generation using SPARTAN (Optional) 20 7 Minimization with new parameters 32 CONTENTS 3 Introduction Molecular dynamics (MD) simulations are a powerful scientific tool used to study a wide variety of systems in atomic detail. From a standard protein simulation, to the use of steered molecular dynamics (SMD), to modelling DNA-protein interactions, there are many useful applications. With the advent of massively parallel simulation programs such as NAMD2, the limits of computational anal- ysis are being pushed even further. Inevitably there comes a time in any molecular modelling scientist’s career when the need to simulate an entirely new molecule or ligand arises. The tech- nique of determining new force field parameters to describe these novel system components therefore becomes an invaluable skill. Determining the correct sys- tem parameters to use in conjunction with the chosen force field is only one important aspect of the process. -
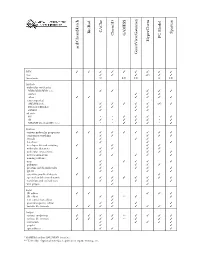
D:\Doc\Workshops\2005 Molecular Modeling\Notebook Pages\Software Comparison\Summary.Wpd
CAChe BioRad Spartan GAMESS Chem3D PC Model HyperChem acd/ChemSketch GaussView/Gaussian WIN TTTT T T T T T mac T T T (T) T T linux/unix U LU LU L LU Methods molecular mechanics MM2/MM3/MM+/etc. T T T T T Amber T T T other TT T T T T semi-empirical AM1/PM3/etc. T T T T T (T) T Extended Hückel T T T T ZINDO T T T ab initio HF * * T T T * T dft T * T T T * T MP2/MP4/G1/G2/CBS-?/etc. * * T T T * T Features various molecular properties T T T T T T T T T conformer searching T T T T T crystals T T T data base T T T developer kit and scripting T T T T molecular dynamics T T T T molecular interactions T T T T movies/animations T T T T T naming software T nmr T T T T T polymers T T T T proteins and biomolecules T T T T T QSAR T T T T scientific graphical objects T T spectral and thermodynamic T T T T T T T T transition and excited state T T T T T web plugin T T Input 2D editor T T T T T 3D editor T T ** T T text conversion editor T protein/sequence editor T T T T various file formats T T T T T T T T Output various renderings T T T T ** T T T T various file formats T T T T ** T T T animation T T T T T graphs T T spreadsheet T T T * GAMESS and/or GAUSSIAN interface ** Text only. -

Development and Application of a Computational Platform for Complex Molecular Design Jaime Rodríguez-Guerra Pedregal
ADVERTIMENT. Lʼaccés als continguts dʼaquesta tesi queda condicionat a lʼacceptació de les condicions dʼús establertes per la següent llicència Creative Commons: http://cat.creativecommons.org/?page_id=184 ADVERTENCIA. El acceso a los contenidos de esta tesis queda condicionado a la aceptación de las condiciones de uso establecidas por la siguiente licencia Creative Commons: http://es.creativecommons.org/blog/licencias/ WARNING. The access to the contents of this doctoral thesis it is limited to the acceptance of the use conditions set by the following Creative Commons license: https://creativecommons.org/licenses/?lang=en Development and Application of a Computational Platform for Complex Molecular Design a dissertation submitted by Jaime Rodríguez-Guerra Pedregal & directed by Prof. Dr. Jean-Didier Maréchal in fulfillment of the requirements for the degree of Doctor of Biotechnology Tutor: Prof. Dr. Jordi Joan Cairó Badillo Department of Chemical, Biological and Environmental Engineering Universitat Autònoma de Barcelona July 2018 Development and Application of a Computational Platform for Complex Molecular Design a dissertation submitted by & recommended for acceptance by advisor Jaime Rodríguez-Guerra Pedregal Prof. Dr. Jean-Didier Maréchal Tutor: Prof. Dr. Jordi Joan Cairó Badillo Department of Chemical, Biological and Environmental Engineering Universitat Autònoma de Barcelona July 2018 ©2018 – Jaime Rodríguez-Guerra Pedregal Licensed as Creative Commons BY-NC-ND Attribution-NonCommercial-NoDerivs In the beginning, there was nothing. And God said «Let there be light». And there was light. There was still nothing, but you could see it a lot better. —WoodyAllen. Development and Application of a Computational Platform for Complex Molecular Design by Jaime Rodríguez-Guerra Pedregal Abstract In this dissertation, a series of novel computational modeling tools is reported. -

Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C
Jaguar 5.5 User Manual Copyright © 2003 Schrödinger, L.L.C. All rights reserved. Schrödinger, FirstDiscovery, Glide, Impact, Jaguar, Liaison, LigPrep, Maestro, Prime, QSite, and QikProp are trademarks of Schrödinger, L.L.C. MacroModel is a registered trademark of Schrödinger, L.L.C. To the maximum extent permitted by applicable law, this publication is provided “as is” without warranty of any kind. This publication may contain trademarks of other companies. October 2003 Contents Chapter 1: Introduction.......................................................................................1 1.1 Conventions Used in This Manual.......................................................................2 1.2 Citing Jaguar in Publications ...............................................................................3 Chapter 2: The Maestro Graphical User Interface...........................................5 2.1 Starting Maestro...................................................................................................5 2.2 The Maestro Main Window .................................................................................7 2.3 Maestro Projects ..................................................................................................7 2.4 Building a Structure.............................................................................................9 2.5 Atom Selection ..................................................................................................10 2.6 Toolbar Controls ................................................................................................11