Characterization of Rifampicin-Resistant Mycobacterium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Muslim 500 2011
The Muslim 500 � 2011 The Muslim The 500 The Muslim 500 � 2011 The Muslim The 500 The Muslim 500The The Muslim � 2011 500———————�——————— THE 500 MOST INFLUENTIAL MUSLIMS ———————�——————— � 2 011 � � THE 500 MOST � INFLUENTIAL MUSLIMS · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · All rights reserved. No part of this book may be repro- The Muslim 500: The 500 Most Influential Muslims duced or utilised in any form or by any means, electronic 2011 (First Edition) or mechanic, inclding photocopying or recording or by any ISBN: 978-9975-428-37-2 information storage and retrieval system, without the prior · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · written permission of the publisher. Views expressed in The Muslim 500 do not necessarily re- Chief Editor: Prof. S. Abdallah Schleifer flect those of RISSC or its advisory board. Researchers: Aftab Ahmed, Samir Ahmed, Zeinab Asfour, Photo of Abdul Hakim Murad provided courtesy of Aiysha Besim Bruncaj, Sulmaan Hanif, Lamya Al-Khraisha, and Malik. Mai Al-Khraisha Image Copyrights: #29 Bazuki Muhammad / Reuters (Page Designed & typeset by: Besim Bruncaj 75); #47 Wang zhou bj / AP (Page 84) Technical consultant: Simon Hart Calligraphy and ornaments throughout the book used courtesy of Irada (http://www.IradaArts.com). Special thanks to: Dr Joseph Lumbard, Amer Hamid, Sun- dus Kelani, Mohammad Husni Naghawai, and Basim Salim. English set in Garamond Premiere -
Cumulative Merit List
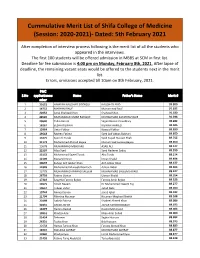
Cummulative Merit List of Shifa College of Medicine (Session: 2020-2021)- Dated: 5th February 2021 After completion of interview process following is the merit list of all the students who appeared in the interviews. The first 100 students will be offered admission in MBBS at SCM in first list. Deadline for fee submission is 4:00 pm on Monday, February 8th, 2021. After lapse of deadline, the remaining vacant seats would be offered to the students next in the merit list. Errors, omissions accepted till 10am on 8th February, 2021. PMC S.No applicationnu Name Father's Name Merit # mber 1 21623 HAMZAH NAUSHAD SIDDIQUI NAUSHAD ABID 93.200 2 14752 MARYAM RAUF Muhammad Rauf 91.632 3 22652 Amal Shahzad Khan Shahzad Khan 91.180 4 28526 MUHAMMAD UMAR RAFIQUE MUHAMMAD ZAFAR RAFIQUE 91.036 5 20640 Talha Rasool Sajjad Rasool Chaudhary 90.282 6 18267 EESHA RIZWAN RIZWAN AHMED 90.045 7 12964 Umar Fakhar Nawaid Fakhar 89.890 8 19618 Raveen Fatima Syed Asif Abbas Bukhari 89.850 9 13475 Syed Ali Turab Syed Sajjad Hussain Shah 89.755 10 31173 Muhammad Ahmed Bajwa Muhammad Farooq Bajwa 89.650 11 13678 MUHAMMAD MEHDI ALI ASAD ALI 89.577 12 21597 Aliza Syed Syed Nadeem Sadiq 89.559 13 15152 Muhammad Sajeel Turab Abu Turab 89.514 14 12109 Manahil Imran Imran Khalid 89.494 15 30637 Zuhayr Arif Jabbar Khan Arif Jabbar Khan 89.477 16 11849 Muhammad Muneeb Warriach Adnan Akbar 89.464 17 13779 MUHAMMAD AMMAD SALEEM MUHAMMAD SALEEM JAVAID 89.427 18 26793 Fatima Usman Usman Khalid 89.354 19 17319 Sayyeda Farooq Bajwa Farooq Amin Bajwa 89.323 20 13675 Alizeh Naeem -

Heralding a New Enlightenment
Conceptualizing the development of personality in children: An analysis of Islamic philosophy and contemporary Western psychology Muhammad Tahir and Stephan Larmar* Abstract: The paper aims to examine the concept of child personality development from the perspectives of Islamic philosophy and contemporary Western psychology. In recent decades, the parental journey associated with the healthy development of children has become increasingly complex and sometimes stressful across all societies and communities of the world. Major world religions and social sciences delineated various aspects and perspectives relating to sound personality development in children. The present article seeks to present the findings of a study that found to give an overview of fundamental principles related to child personality development drawn from Islamic philosophy and contemporary Western psychology. Drawing on these two perspectives, the paper seeks to explain personality development, highlighting both similarities and differences associated with these perspectives. The study employed qualitative content analysis to explore relevant data from the Qur’anic verses and Prophetic traditions, as well as theoretical studies and empirical research of psychology. The research findings predominantly highlight an integrated approach towards child personality development as framed within the perspective of Islamic philosophy and contemporary Western psychological understandings. The paper serves to link Islamic thought to contemporary Western psychological aspects -

Conferment of Pakistan Civil Awards - 14Th August, 2020
F. No. 1/1/2020-Awards-I GOVERNMENT OF PAKISTAN CABINET SECRETARIAT (CABINET DIVISION) ***** PRESS RELEASE CONFERMENT OF PAKISTAN CIVIL AWARDS - 14TH AUGUST, 2020 On the occasion of Independence Day, 14th August, 2020, the President of the Islamic Republic of Pakistan has been pleased to confer the following ‘Pakistan Civil Awards’ on citizens of Pakistan as well as Foreign Nationals for showing excellence and courage in their respective fields. The investiture ceremony of these awards will take place on Pakistan Day, 23rd March, 2021:- S. No. Name of Awardee Field 1 2 3 I. NISHAN-I-IMTIAZ 1 Mr. Sadeqain Naqvi Arts (Painting/Sculpture) 2 Prof. Shakir Ali Arts (Painting) 3 Mr. Zahoor ul Haq (Late) Arts (Painting/ Sculpture) 4 Ms. Abida Parveen Arts (Singing) 5 Dr. Jameel Jalibi Literature Muhammad Jameel Khan (Late) (Critic/Historian) (Sindh) 6 Mr. Ahmad Faraz (Late) Literature (Poetry) (Khyber Pakhtunkhwa) II. HILAL-I-IMTIAZ 7 Prof. Dr. Anwar ul Hassan Gillani Science (Pharmaceutical (Sindh) Sciences) 8 Dr. Asif Mahmood Jah Public Service (Punjab) III. HILAL-I-QUAID-I-AZAM 9 Mr. Jack Ma Services to Pakistan (China) IV. SITARA-I-PAKISTAN 10 Mr. Kyu Jeong Lee Services to Pakistan (Korea) 11 Ms. Salma Ataullahjan Services to Pakistan (Canada) V. SITARA-I-SHUJA’AT 12 Mr. Jawwad Qamar Gallantry (Punjab) 13 Ms. Safia (Shaheed) Gallantry (Khyber Pakhtunkhwa) 14 Mr. Hayatullah Gallantry (Khyber Pakhtunkhwa) 15 Malik Sardar Khan (Shaheed) Gallantry (Khyber Pakhtunkhwa) 16 Mr. Mumtaz Khan Dawar (Shaheed) Gallantry (Khyber Pakhtunkhwa) 17 Mr. Hayat Ullah Khan Dawar Hurmaz Gallantry (Shaheed) (Khyber Pakhtunkhwa) 18 Malik Muhammad Niaz Khan (Shaheed) Gallantry (Khyber Pakhtunkhwa) 19 Sepoy Akhtar Khan (Shaheed) Gallantry (Khyber Pakhtunkhwa) 20 Mr. -

Effects of Lysine Levels with Digestible Amino Acids on Growth Performance, Carcass Evaluation, Productive and Reproductive Traits of Aseel Chicken
EFFECTS OF LYSINE LEVELS WITH DIGESTIBLE AMINO ACIDS ON GROWTH PERFORMANCE, CARCASS EVALUATION, PRODUCTIVE AND REPRODUCTIVE TRAITS OF ASEEL CHICKEN MUNAWAR HUSSAIN 2014-VA-509 A THESIS SUBMITTED IN THE PARTIAL FULFILLMENT OF THE REQUIREMENT FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN POULTRY PRODUCTION UNIVERSITY OF VETERINARY AND ANIMAL SCIENCES LAHORE, PAKISTAN 2018 To, The Controller of Examinations, University of Veterinary and Animal Sciences, Lahore. We, the Supervisory Committee, certify that the contents and form of the thesis, submitted by Munawar Hussain, Reg. # 2014-VA-509 have been found satisfactory and recommend that it be processed for the evaluation by the External Examiner (s) for award of the Degree. Supervisor ___________________________________________ (Prof. Dr. Athar Mahmud) Member ___________________________________________ (Dr. Jibran Hussain) Member ___________________________________________ (Dr. Shafqat Nawaz Qaisrani) DEDICATION This work is dedicated to My Beloved Parents& Family My Teachers especially Prof. Dr. Muhammad Akram i ACKNOWLEDGEMENTS All praises and thanks to ALMIGHTY ALLAH, the Lord of the worlds, the Omnipotent, the most Beneficent, the Merciful and the Gracious. Who is the creator of this world and heavens, the lord of the Day of Judgment, who is the entire source of knowledge and wisdom and endowed it to the mankind. The work described in this thesis was made possible by contributions from various individuals and institutions to whom I am indebted. I thank all those who, in different ways, have walked beside me along the way; offering support and encouragement, challenging my thinking, and teaching me to consider alternative views. The author would like to express his deep appreciation and thanks to my supervisor Prof. -

Ottomans-Safavids-Mughals:Shared Knowledge and Connective Systems Francis Robinson the Boundaries of Modern Nation-States An
.. Ottomans-Safavids-Mughals:Shared knowledge and connective Systems Francis Robinson The boundaries of modern nation-states and the blinkered view of area studies scholarship have tended to obscure both important areas of shared experience and significant systems of connection between the Middle East and South Asia. If this is true of the structural characteristics of the Ottoman, Safavid and Mughal empires, of the ways in which their local, regional and imperial systems were articulated, and if this is also true of their commercial organisation and techniques of trade, this is no less true of the content of their systems of formal learning, of the nature of their major sources of esoteric understanding, and of the ways in which they were linked by the connective systems of learned and holy men. By comparing the curriculums taught in the madrasas of the three empires up to the end of the seventeenth century we will aim to reveal the differing balances maintained between the transmitted subjects (`ulum-i naqliyya/manqulat) and the rational subjects (`ulum-i `aqliyya/ma`qulat). We will also examine the extent to which madrasas adopted the same texts, and even used the same commentaries and annotations. That there were shared texts and commentaries was a consequence of the travels of scholars throughout the region. Often they journeyed in search of knowledge, but they did so too in search of both patrons to sustain their work and safety from oppression. The paths they 2 followed were the channels along which ideas came to be shared; the centres at which they congregated were the places from which ideas were broadcast. -

Dogmatic Approaches of Qur'ān Translators
Dogmatic Approaches of Qur’ān Translators: Linguistic and Theological Issues Somia Qudah-Refai Submitted in accordance with the requirements for the degree of Doctor of Philosophy School of Modern Languages and Cultures University of Leeds May 2014 Intellectual Property and Publication Statements The candidate confirms that the work submitted is her own and that appropriate credit has been given where reference has been made to the work of others. This copy has been supplied on the understanding that it is copyright material and that no quotation from the thesis may be published without proper acknowledgement University of Leeds Somia Qudah-Refai ii ‘Lord, inspire me to be thankful for the blessings You have granted me and my parents, and to do good deeds that please You; admit me by Your grace into the ranks of Your righteous servants’ (Qur’ān, 27:19). This work is dedicated to my beloved parents, Dr. Abdul-Hameed and Mrs. Nedal Al-Qudah, for their endless love and everlasting prayers. You contributed to my life far more than what I will ever be able to thank you for, to you I say: Jazakum Allah Khairan iii Acknowledgements All praise is to God for enabling me to fulfil the requirements of this study. My sincere gratitude goes to my initial supervisor Prof. Hussein Abdul-Raof and my current supervisor Prof. James Dickins. Prof. Abdul-Raof provided me with guidance and advice when I was establishing the research project and continued to do so during his time in the University of Leeds. I would not have been able to finish this work without the valuable advice, guidance, assistance and encouragement of Prof. -

Election to the Provincial Assembly of the Sindh General Election 2007-2008
LIST OF CONTESTING CANDIDATES ELECTION TO THE PROVINCIAL ASSEMBLY OF THE SINDH GENERAL ELECTION 2007-2008 NO. AND NAME OF PARTY S NO NAME OF CANDIDATE CONSTITUENCY AFFILIATION 1 23 5 1 Agha Iftikhar-u-Din. INDEPENDENT 2 Aftab Alam. INDEPENDENT 3 Imtiaz Hussain ST 4 M. Hanif Memon. INDEPENDENT 5 Tanveer Ahmad Rajput INDEPENDENT 6 Hamid Mehmood Faizi. INDEPENDENT 7 Dr.Nasrullah Baloch. PPPP 8 Zahid Hussain Shoro. PML-N 9 Siraj Ahmed Khan INDEPENDENT 10 Saeed Iqbal. MQM PS-1, Sukkur-I 11 Atif Ilyas Khan Advocate. INDEPENDENT 12 Abdul Jabbar Solangi INDEPENDENT 13 Abdul Mateen Bandhani. MMA 14 Muhammad Arshad Mughul. INDEPENDENT 15 Muhammad Iqbal. INDEPENDENT 16 Muhammad Saeed Mughul INDEPENDENT 17 Muhammad Saleem Khan INDEPENDENT 18 Muhammad Yousuf Umar Shaikh INDEPENDENT 19 Mushtaque Ahmad Mirani. INDEPENDENT 20 Muhammad Asad Thanvi JUI-S 21 Naheed Baji INDEPENDENT 1 Agha Saifullah Pathan. INDEPENDENT 2 Anwar Ahmad Khan Mahar. PPPP 3 Dilawar Khan INDEPENDENT 4 Sardar Mir Yakoob Ali Shah SUP 5 Syed Farrukh Ahmad Shah. INDEPENDENT 6 Syed Muhammad Irfan Ali Shah. ST PS-2, Sukkur-cum- 7 Tariq Sulleman INDEPENDENT Shikarpur (Old Sukkur-II) 8 Abdul Jabbar Solangi INDEPENDENT 9 Abdul Ghani. MMA 10 Abdul Mujeeb Pirzada NPP 11 Gul Hassan Khoso PML-N DESIGNED AND DEVELOPED BY IT WING PEC SINDH Page 1 NO. AND NAME OF PARTY S NO NAME OF CANDIDATE CONSTITUENCY AFFILIATION 12 Muhammad Ayoub INDEPENDENT 13 Muhammad Ali Shaikh INDEPENDENT 14 Munawar Ali Choohan. MQM 1 Badaruddin Samejo INDEPENDENT 2 Jam Ikramullah Khan Dharejo INDEPENDENT 3 Jam Saifullah -

36Th Session 1951)
Pakistan Engineering Congress in Retrospect (1912 – 2012) Centenary Celebration 659 CONGRESS IN PICTURES PUNJAB ENGINEERING CONGRESS (36TH SESSION 1951) 6th Row Standing: Nisar Ahmed Malik; Abdul Mannan Sheikh; M. Aslam Mufti; M. A. Malik; R. R. Shariff 5th Row Standing: M. Ahmed; N. M. Chaudri; Mazhar Munir; Khalid Faruq Akbar; M. I. Chishti; M. Masud Ahmed; A. W. Khokhar; Mazhar-ul-Haq; Latif Hussain; Muhammad Rashid Vehra; Kamal Mustafa; Ashfaq Hasan; Ferozeuddin Ahmad; S. I. Ahmed; M. Shamim Ahmed; S. M. Niaz; Monawar Ali; Syed Irfan Ahmed; Mir Sadaqat Hassan; Muhammad Ashraf Cheema; G. M. Sheikh; Kh. Maqbool Ahmad; Mian Bashir Ahmed. 4th Row Standing: Muhammad Saadat Ali; Mian Muhammad Hayat; G. M. Subhani; M. A. Hamid Rahmani; Sh. Zahoor ul Haq; Khawaja Saleemud Din; S Ahmed Ali Shah; Abdul Hamid; S. A. Majeed; Ch. Muhammad Ali; M. T. Shah; A. Khalique; Malik Aman Ullah; Syed Iftikhar Ahmed; Muhammad Hanif; Shafique Ahmed; M A. Rashid; Fazal Karim; Shaukat Omar; Aziz Omar; M. A. Hafiz Khan; H. S. Zaidi; Ch. Abdul Hamid; Syed Mahdi Shah; Asrar Qureshy. 3rd Row Standing: S.A. Qureshi; S. M. Serajuz Zaman; S. Hamiud Din Ahmed; Sh. Abdur Rawoof; D. M Khanzada; M. H. Siddiqi; Rab Nawaz Batra; Sh. Mukhtar Mahmood; S. Jaffar Hussain; Malik N. M. Khan; Jameel A. Parvez; M. Faizanul Haq; H. J. Asar; F. Rahman: M. Saeed Ahmed; S. I. A. Shah; Muhammad Akram; M. M. Haque; M. S. Minhas; Yusuf Ali Siddiqi; S. S. Kirmani; I. A. S. Bokhari. 2nd Row Standing: Muhammad Aslam Butt; Ch. Abdul Azjz; Mian Muhammad Yunas; S. -

Naqshbandi Sufi, Persian Poet
ABD AL-RAHMAN JAMI: “NAQSHBANDI SUFI, PERSIAN POET A Dissertation Presented in Partial Fulfillment of the Requirement for The Degree Doctor of Philosophy in the Graduate School of the Ohio State University By Farah Fatima Golparvaran Shadchehr, M.A. The Ohio State University 2008 Approved by Professor Stephen Dale, Advisor Professor Dick Davis Professor Joseph Zeidan ____________________ Advisor Graduate Program in History Copyright by Farah Shadchehr 2008 ABSTRACT The era of the Timurids, the dynasty that ruled Transoxiana, Iran, and Afghanistan from 1370 to 1506 had a profound cultural and artistic impact on the history of Central Asia, the Ottoman Empire, and Mughal India in the early modern era. While Timurid fine art such as miniature painting has been extensively studied, the literary production of the era has not been fully explored. Abd al-Rahman Jami (817/1414- 898/1492), the most renowned poet of the Timurids, is among those Timurid poets who have not been methodically studied in Iran and the West. Although, Jami was recognized by his contemporaries as a major authority in several disciplines, such as science, philosophy, astronomy, music, art, and most important of all poetry, he has yet not been entirely acknowledged in the post Timurid era. This dissertation highlights the significant contribution of Jami, the great poet and Sufi thinker of the fifteenth century, who is regarded as the last great classical poet of Persian literature. It discusses his influence on Persian literature, his central role in the Naqshbandi Order, and his input in clarifying Ibn Arabi's thought. Jami spent most of his life in Herat, the main center for artistic ability and aptitude in the fifteenth century; the city where Jami grew up, studied, flourished and produced a variety of prose and poetry. -

Afghanistan INDIVIDUALS
CONSOLIDATED LIST OF FINANCIAL SANCTIONS TARGETS IN THE UK Last Updated:01/02/2021 Status: Asset Freeze Targets REGIME: Afghanistan INDIVIDUALS 1. Name 6: ABBASIN 1: ABDUL AZIZ 2: n/a 3: n/a 4: n/a 5: n/a. DOB: --/--/1969. POB: Sheykhan village, Pirkowti Area, Orgun District, Paktika Province, Afghanistan a.k.a: MAHSUD, Abdul Aziz Other Information: (UK Sanctions List Ref):AFG0121 (UN Ref): TAi.155 (Further Identifiying Information):Key commander in the Haqqani Network (TAe.012) under Sirajuddin Jallaloudine Haqqani (TAi.144). Taliban Shadow Governor for Orgun District, Paktika Province as of early 2010. Operated a training camp for non Afghan fighters in Paktika Province. Has been involved in the transport of weapons to Afghanistan. INTERPOL-UN Security Council Special Notice web link: https://www.interpol.int/en/How-we- work/Notices/View-UN-Notices-Individuals click here. Listed on: 21/10/2011 Last Updated: 01/02/2021 Group ID: 12156. 2. Name 6: ABDUL AHAD 1: AZIZIRAHMAN 2: n/a 3: n/a 4: n/a 5: n/a. Title: Mr DOB: --/--/1972. POB: Shega District, Kandahar Province, Afghanistan Nationality: Afghan National Identification no: 44323 (Afghan) (tazkira) Position: Third Secretary, Taliban Embassy, Abu Dhabi, United Arab Emirates Other Information: (UK Sanctions List Ref):AFG0094 (UN Ref): TAi.121 (Further Identifiying Information): Belongs to Hotak tribe. Review pursuant to Security Council resolution 1822 (2008) was concluded on 29 Jul. 2010. INTERPOL-UN Security Council Special Notice web link: https://www.interpol.int/en/How-we-work/ Notices/View-UN-Notices-Individuals click here. Listed on: 23/02/2001 Last Updated: 01/02/2021 Group ID: 7055. -

Exploring Qur'anic Stance on Mental Health
Exploring Qur’anic Stance on Mental Health: An Analytical Review University of Wah Journal of Social Sciences Volume 2, Issue 2, Dec 2019, pp. 46-71 Exploring Qur’anic Stance on Mental Health: An Analytical Review Dr. Muhammad Tahir1, Dr. Sami Ullah Zubairi2 Article History: ABSTRACT Received: In recent decades, there is a massive increase in mental health issues 09 Apr 2019 worldwide. Muslim communities seemingly facing mental health issues at an alarming rate. Traditionally, mental health is an important issue among public Accepted: health issues. Therefore, religions and social sciences reflect upon its various 16 Oct 2019 aspects. Necessarily, religion and mental health have had deep connections and relationships throughout human history. All the Abraham religions provide awareness and guidance for mental health issues from a religious domain. Thus, mental health has become a subject of more attention for religious scholars too, which creates a current need for addressing this issue from an Islamic perspective. Islam, being a complete code of life for Muslims, provides a holistic overview of mental health issues for the development of sound masses in society. Islamic teachings describe a vibrant coping mechanism to deal with mental illnesses, psychological problems, and uncertain conditions of life. The present study draws attention specifically on mental health and well-being from the Qur’anic discourse to present possible solutions within an Islamic domain. The presented understanding of Qur’anic stance on mental health incorporates with human needs and requirements to provide a practical framework for the improvement of general health conditions in the contemporary Muslim world, multi-cultural societies, and the multi-religious Western world.