Synthesis of Asymmetrically Substituted Scyllo-Inositol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inositol Safety: Clinical Evidences
European Review for Medical and Pharmacological Sciences 2011; 15: 931-936 Inositol safety: clinical evidences G. CARLOMAGNO, V. UNFER AGUNCO Obstetrics & Gynecology Center, Rome (Italy) Abstract. – Myo-inositol is a six carbon ent required by the human cells for the growth cyclitol that contains five equatorial and one axi- and survival in the culture. In humans and other al hydroxyl groups. Myo-inositol has been classi- species, Myo-inositol can be converted to either fied as an insulin sensitizing agent and it is L- or D-chiro-inositol by epimerases. Early stud- commonly used in the treatment of the Polycys- tic Ovary Syndrome (PCOS). However, despite ies showed that inositol urinary clearance was al- its wide clinical use, there is still scarce informa- tered in type 2 diabetes patients, the next step tion on the myo-inositol safety and/or side ef- was to link impaired inositol clearance with in- fects. The aim of the present review was to sum- sulin resistance (for a review see1). Because of marize and discuss available data on the myo-in- these properties, inositol have been classified as ositol safety both in non-clinical and clinical set- “insulin sensitizing agent”2. tings. The main outcome was that only the highest In the recent years, inositol has found more dose of myo-inositol (12 g/day) induced mild and more space in the reproductive clinical prac- gastrointestinal side effects such as nausea, fla- tice3-6. Indeed, since the main therapy for Poly- tus and diarrhea. The severity of side effects did cystic Ovary Syndrome (PCOS) is the use of in- not increase with the dosage. -

Revisions to USP 31–NF 26, Second Supplement
Revisions to USP 31–NF 26, Second Supplement (published May 2008) Published April 2008 General Chapters Monographs:A–C D–N O–S T–Z Monograph Title Section Head Scientific Liaison DANTROLENE SODIUM Dissolution <711> CAPSULES PF 33(4) Pg. 645 DEHYDROACETIC ACID PF 33(4) Title Pg. 703 DEHYDROACETIC ACID PF 33(4) Chemical Info Pg. 703 DEHYDROACETIC ACID PF 33(4) Definition Pg. 703 DEHYDROACETIC ACID PF 33(4) Packaging and storage Pg. 703 DEHYDROACETIC ACID PF 33(4) USP Reference standards <11> Pg. 703 DEHYDROACETIC ACID PF 33(4) Identification Pg. 703 DEHYDROACETIC ACID PF 33(4) Heavy Metals, Method II <231> Pg. 703 DEHYDROACETIC ACID PF 33(4) Loss on drying <731> Pg. 703 DEHYDROACETIC ACID PF 33(4) Melting range, Class I <741> Pg. 703 DEHYDROACETIC ACID PF 33(4) Residue on ignition <281> Pg. 703 DEHYDROACETIC ACID PF 33(4) Assay Pg. 703 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Title 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Definition 784 file:///C|/Documents%20and%20Settings/rwt/Desktop/revisions/usp31nf26secondSupplement03.html[4/26/2011 1:18:07 PM] DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Packaging and storage 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Labeling 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. USP Reference stadards 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Identification 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Uniformity of dosage units 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. Loss on drying 784 DIDANOSINE TABLETS FOR ORAL SUSPENSION PF 32(3) Pg. -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

Genetic Control of Apigenin Di-C-Glycoside Biosynthesis in Bread Wheat Grain and Their Role
Genetic control of Apigenin di-C-glycoside biosynthesis in bread wheat grain and their role as yellow pigments of Asian alkaline noodles Submitted by Grace Yasmein Wijaya This thesis is submitted to Faculty of Sciences in fulfilment of the requirements for the degree Doctor of Philosophy Faculty of Science The University of Adelaide November 2012 This book is An Answer to Prayers of a Long list of Believers I have been very blessed and loved with all of your spiritual supports, for His guidance and protections, and sincerely will not have enough to thank you all..... May you all be blessed and loved, too As I have been always, yasmein Table of Content Table of Content i List of Tables x List of Figures xiii List of Supplemental Materials xxv Summary xxx Statement of Authorship xxxiv List of Publications xxxv Acknowledgement xxxvi List of Abbreviations xxxix Chapter I: General Introduction 1 1.1 Background 1 1.2 Knowledge Gap 2 1.3 Structure of thesis 2 Chapter II: Literature review 4 2.1 Asian noodles as one of the major end-products of Australian bread wheat 4 2.2 Yellow colour of YAN 5 2.2.1 Natural Compounds that contribute to the yellow colour of YAN 5 2.2.1.1 Types of natural compounds that contributes to the yellow colour of YAN and their roles in plants and human health 5 2.2.1.2 Contribution of xanthophylls and ACGs to the yellow colour of alkaline noodles 8 2.2.2 Factors influencing the measurement of the yellowness of noodles and the content of xanthophyll and ACG in wheat grain 10 i 2.2.3 The amount, tissue location and composition -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -
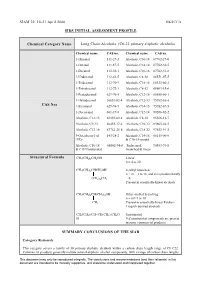
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Inositol (Powder) Introduced 2008
Product Information Sheet – March 2016 Inositol (powder) Introduced 2008 What Is It? Inositol (powder) Inositol is a component of the B-complex family. It supports healthy two scoops (approximately 4.2 g) contain v central nervous system function, including emotional wellness, healthy mood and behavior. Additionally, it may promote ovarian health.* inositol (as myo-inositol) .................................................................................4.2 g serving size: approximately 4.2 g (2 scoops) Uses For Inositol (powder) servings per container: 60 Emotional Wellness: Myo-inositol is the primary form of inositol 2 scoops, 1–2 times daily, with or between meals, or as directed found in the central nervous system. It plays an important role in cell by a health professional. membrane formation and serves as part of the phosphatidylinositol second messenger system, supporting serotonin, norepinenephrine and cholinergic receptor function. As a result, inositol may support healthy mood, emotional wellness and behavior, and ease occasional nervous tension.* Ovarian Function: Research suggests that myo-inositol may help to support healthy ovulatory activity, ovarian function and reproductive system function.* What Is The Source? Myo-inositol is derived from rice bran. Recommendations Pure Encapsulations® recommends 2 scoops, 1–2 times daily, with or between meals, or as directed by a health professional. Are There Any Potential Side Effects Or Precautions? Rarely, inositol has been associated with nausea, fatigue, headache or dizziness. If pregnant or lactating, consult your physician before taking this product. Are There Any Potential Drug Interactions? At this time, there are no known adverse reactions when taken in conjunction with medications. Consult your physician for more information. *These statements have not been evaluated by the Food and Drug Administration. -

WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 36/84 (2006.01) A61K 31/5513 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/045 (2006.01) A61P 31/22 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/522 (2006.01) A61K 45/06 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/NL20 14/050780 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 13 November 2014 (13.1 1.2014) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/903,430 13 November 2013 (13. 11.2013) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: RJG DEVELOPMENTS B.V. -

Transcription 12.01.12
Lecture 2B • 01/12/12 We covered three different reactions for converting alcohols into leaving groups. One was to turn an alcohol into an alkyl chloride, that was using thionyl chloride. Second reaction was using tosyl chloride; the primary difference between those two reactions is one of stereochemistry. An inversion of stereochemistry does occur if you use thionyl chloride, because it does affect the carbon-oxygen bond, but because forming a tosylate does not touch the carbon-oxygen bond, only the oxygen- hydrogen bond, there’s no change in stereochemistry there. We then saw phosphorus tribromide that reacts very similarly to the thionyl chloride; you get an alkyl bromide instead, but it also has inversion of configuration. The last reaction is not a new mechanism, it is just an Sn2 reaction, it’s called the Finkelstein reaction. Really this works, in a sense, off of Le Châtelier’s principle. In solution, in theory, sodium iodide can displace bromide, but sodium bromide can displace iodide, so you can have an Sn2 reaction that goes back and forth and back and forth and back and forth. Except, sodium iodide is somewhat soluble in acetone, while sodium chloride and sodium bromide are not. So, in fact, one of the things that we get out of this as a by-product is sodium bromide, which, again, is not soluble in acetone and therefore precipitates out and is no longer part of the reaction mixture, so there’s no reverse reaction possible. Because of this solubility trick, it allows this reaction to be pulled forward, which means you can get the alkyl iodide. -

Synthesis of Mannosylglucosaminylinositol
Proc. Nati. Acad. Sci. USA Vol. 89, pp. 6025-6029, July 1992 Medical Sciences Synthesis of mannosylglucosaminylinositol phospholipids in normal but not paroxysmal nocturnal hemoglobinuria cells (complement/decay-accelerating factor/CD59) SHINICHI HIROSE*, LAKSHMESWARI RAVI*, GREGORY M. PRINCE*, MELVIN G. ROSENFELDt, ROBERT SILBERf, STEVEN W. ANDRESEN§, SANDRA V. HAZRA¶, AND M. EDWARD MEDOF*II *Institute of Pathology, Case Western Reserve University, Cleveland, OH 44106; Departments of tCell Biology and *Medicine, New York University Medical Center, New York, NY 10016; §Cleveland Clinic Foundation, Cleveland, OH 44106; and lAkron General Medical Center, Akron, OH 44307 Communicated by Frederick C. Robbins, November 29, 1991 (received for review September 12, 1991) ABSTRACT To identify mannosyl (Man)-containing inter- have been identified, information concerning the biochemical mediates ofthe human glycoinositol phospholipid (GPI) anchor pathway(s) responsible for their intracellular GPI-anchor pathway and examine their expression in paroxysmal nocturnal assembly and protein incorporation is only beginning to hemoglobinuria (PNH), mannolipid products deriving from in emerge. vitro guanosine diphosphate [3HJMan labeling of HeLa cell Among human GPI-anchored proteins are the decay- microsomes were characterized. The defined GPI species were accelerating factor (DAF) and CD59, membrane-associated correlated with products deriving from in vivo [3HJMan label- regulators of the complement cascade which protect host ing of normal and (GPI-anchor defective) affected leukocytes. tissues from autologous complement attack (reviewed in refs. In vitro analyses in HeLa cells showed dolichol-phosphoryl 2-4). Deficient expression of these proteins underlies an (Dol-P)-[3H]Man and a spectrum of [3H]Man lipids exhibiting acquired hemolytic disorder termed paroxysmal nocturnal TLC mobilities approximating those of Trypanosoma brucei hemoglobinuria (PNH), and the lesion responsible for the (Tryp) GPI precursors. -

Ion Channels
UC Davis UC Davis Previously Published Works Title THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels. Permalink https://escholarship.org/uc/item/1442g5hg Journal British journal of pharmacology, 176 Suppl 1(S1) ISSN 0007-1188 Authors Alexander, Stephen PH Mathie, Alistair Peters, John A et al. Publication Date 2019-12-01 DOI 10.1111/bph.14749 License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California S.P.H. Alexander et al. The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British Journal of Pharmacology (2019) 176, S142–S228 THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Ion channels Stephen PH Alexander1 , Alistair Mathie2 ,JohnAPeters3 , Emma L Veale2 , Jörg Striessnig4 , Eamonn Kelly5, Jane F Armstrong6 , Elena Faccenda6 ,SimonDHarding6 ,AdamJPawson6 , Joanna L Sharman6 , Christopher Southan6 , Jamie A Davies6 and CGTP Collaborators 1School of Life Sciences, University of Nottingham Medical School, Nottingham, NG7 2UH, UK 2Medway School of Pharmacy, The Universities of Greenwich and Kent at Medway, Anson Building, Central Avenue, Chatham Maritime, Chatham, Kent, ME4 4TB, UK 3Neuroscience Division, Medical Education Institute, Ninewells Hospital and Medical School, University of Dundee, Dundee, DD1 9SY, UK 4Pharmacology and Toxicology, Institute of Pharmacy, University of Innsbruck, A-6020 Innsbruck, Austria 5School of Physiology, Pharmacology and Neuroscience, University of Bristol, Bristol, BS8 1TD, UK 6Centre for Discovery Brain Science, University of Edinburgh, Edinburgh, EH8 9XD, UK Abstract The Concise Guide to PHARMACOLOGY 2019/20 is the fourth in this series of biennial publications. The Concise Guide provides concise overviews of the key properties of nearly 1800 human drug targets with an emphasis on selective pharmacology (where available), plus links to the open access knowledgebase source of drug targets and their ligands (www.guidetopharmacology.org), which provides more detailed views of target and ligand properties. -

Reactions of Alcohols & Ethers 1
REACTIONS OF ALCOHOLS & ETHERS 1. Combustion (Extreme Oxidation) alcohol + oxygen carbon dioxide + water 2 CH3CH2OH + 6 O2 4 CO2 + 6 H2O 2. Elimination (Dehydration) ° alcohol H 2 S O 4/ 1 0 0 C alkene + water H2SO4/100 ° C CH3CH2CH2OH CH3CH=CH2 + H2O 3. Condensation ° excess alcohol H 2 S O 4 / 1 4 0 C ether + water H2SO4/140 ° C 2 CH3CH2OH CH3CH2OCH2CH3 + H2O 4. Substitution Lucas Reagent alcohol + hydrogen halide Z n C l2 alkyl halide + water ZnCl2 CH3CH2OH + HCl CH3CH2Cl + H2O • This reaction with the Lucas Reagent (ZnCl2) is a qualitative test for the different types of alcohols because the rate of the reaction differs greatly for a primary, secondary and tertiary alcohol. • The difference in rates is due to the solubility of the resulting alkyl halides • Tertiary Alcohol→ turns cloudy immediately (the alkyl halide is not soluble in water and precipitates out) • Secondary Alcohol → turns cloudy after 5 minutes • Primary Alcohol → takes much longer than 5 minutes to turn cloudy 5. Oxidation • Uses an oxidizing agent such as potassium permanganate (KMnO4) or potassium dichromate (K2Cr2O7). • This reaction can also be used as a qualitative test for the different types of alcohols because there is a distinct colour change. dichromate → chromium 3+ (orange) → (green) permanganate → manganese (IV) oxide (purple) → (brown) Tertiary Alcohol not oxidized under normal conditions CH3 KMnO4 H3C C OH NO REACTION K2Cr2O7 CH3 tertbutyl alcohol Secondary Alcohol ketone + hydrogen ions H O KMnO4 H3C C CH3 + K2Cr2O7 C + 2 H H3C CH3 OH propanone 2-propanol Primary Alcohol aldehyde + water carboxylic acid + hydrogen ions O KMnO4 O H H KMnO H H 4 + CH3CH2CH2OH C C C H C C C H + 2 H K2Cr2O7 + H2O K Cr O H H H 2 2 7 HO H H 1-propanol propanal propanoic acid 6.