Differential Expression of Neuronal Genes Defines Subtypes Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

MCG10 (PCBP4) (NM 033008) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP300749 MCG10 (PCBP4) (NM_033008) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human poly(rC) binding protein 4 (PCBP4), transcript variant 3 Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 41.3 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_127501 Locus ID: 57060 UniProt ID: P57723, A0A024R2Y0 RefSeq Size: 2040 Cytogenetics: 3p21.2 RefSeq ORF: 1209 Synonyms: CBP; LIP4; MCG10 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 MCG10 (PCBP4) (NM_033008) Human Recombinant Protein – TP300749 Summary: This gene encodes a member of the KH-domain protein subfamily. Proteins of this subfamily, also referred to as alpha-CPs, bind to RNA with a specificity for C-rich pyrimidine regions. Alpha-CPs play important roles in post-transcriptional activities and have different cellular distributions. -

Downloads/ (Accessed on 17 January 2020)
cells Review Novel Approaches for Identifying the Molecular Background of Schizophrenia Arkadiy K. Golov 1,2,*, Nikolay V. Kondratyev 1 , George P. Kostyuk 3 and Vera E. Golimbet 1 1 Mental Health Research Center, 34 Kashirskoye shosse, 115522 Moscow, Russian; [email protected] (N.V.K.); [email protected] (V.E.G.) 2 Institute of Gene Biology, Russian Academy of Sciences, 34/5 Vavilova Street, 119334 Moscow, Russian 3 Alekseev Psychiatric Clinical Hospital No. 1, 2 Zagorodnoye shosse, 115191 Moscow, Russian; [email protected] * Correspondence: [email protected] Received: 5 November 2019; Accepted: 16 January 2020; Published: 18 January 2020 Abstract: Recent advances in psychiatric genetics have led to the discovery of dozens of genomic loci associated with schizophrenia. However, a gap exists between the detection of genetic associations and understanding the underlying molecular mechanisms. This review describes the basic approaches used in the so-called post-GWAS studies to generate biological interpretation of the existing population genetic data, including both molecular (creation and analysis of knockout animals, exploration of the transcriptional effects of common variants in human brain cells) and computational (fine-mapping of causal variability, gene set enrichment analysis, partitioned heritability analysis) methods. The results of the crucial studies, in which these approaches were used to uncover the molecular and neurobiological basis of the disease, are also reported. Keywords: schizophrenia; GWAS; causal genetic variants; enhancers; brain epigenomics; genome/epigenome editing 1. Introduction Schizophrenia is a severe mental illness that affects between 0.5% and 0.7% of the human population [1]. Both environmental and genetic factors are thought to be involved in its pathogenesis, with genetic factors playing a key role in disease risk, as the heritability of schizophrenia is estimated to be 70–85% [2,3]. -

TRPV4: a Physio and Pathophysiologically Significant Ion Channel
International Journal of Molecular Sciences Review TRPV4: A Physio and Pathophysiologically Significant Ion Channel Tamara Rosenbaum 1,* , Miguel Benítez-Angeles 1, Raúl Sánchez-Hernández 1, Sara Luz Morales-Lázaro 1, Marcia Hiriart 1 , Luis Eduardo Morales-Buenrostro 2 and Francisco Torres-Quiroz 3 1 Departamento de Neurociencia Cognitiva, División Neurociencias, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Mexico City 04510, Mexico; [email protected] (M.B.-A.); [email protected] (R.S.-H.); [email protected] (S.L.M.-L.); [email protected] (M.H.) 2 Departamento de Nefrología y Metabolismo Mineral, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City 14080, Mexico; [email protected] 3 Departamento de Bioquímica y Biología Estructural, División Investigación Básica, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Mexico City 04510, Mexico; [email protected] * Correspondence: [email protected]; Tel.: +52-555-622-56-24; Fax: +52-555-622-56-07 Received: 3 May 2020; Accepted: 24 May 2020; Published: 28 May 2020 Abstract: Transient Receptor Potential (TRP) channels are a family of ion channels whose members are distributed among all kinds of animals, from invertebrates to vertebrates. The importance of these molecules is exemplified by the variety of physiological roles they play. Perhaps, the most extensively studied member of this family is the TRPV1 ion channel; nonetheless, the activity of TRPV4 has been associated to several physio and pathophysiological processes, and its dysfunction can lead to severe consequences. Several lines of evidence derived from animal models and even clinical trials in humans highlight TRPV4 as a therapeutic target and as a protein that will receive even more attention in the near future, as will be reviewed here. -

Spermatozoal Gene KO Studies for the Highly Present Paternal Transcripts
Spermatozoal gene KO studies for the highly present paternal Potential maternal interactions Conclusions of the KO studies on the maternal gene transcripts detected by GeneMANIA candidates for interaction with the paternal Fbxo2 Selective cochlear degeneration in mice lacking Cul1, Fbxl2, Fbxl3, Fbxl5, Cul-1 KO causes early embryonic lethality at E6.5 before Fbxo2 Fbxo34, Fbxo5, Itgb1, Rbx1, the onset of gastrulation http://www.jneurosci.org/content/27/19/5163.full Skp1a http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3641602/. Loss of Cul1 results in early embryonic lethality and Another KO study showed the following: Loss of dysregulation of cyclin E F-box only protein 2 (Fbxo2) disrupts levels and http://www.ncbi.nlm.nih.gov/pubmed/10508527?dopt=Abs localization of select NMDA receptor subunits, tract. and promotes aberrant synaptic connectivity http://www.ncbi.nlm.nih.gov/pubmed/25878288. Fbxo5 (or Emi1): Regulates early mitosis. KO studies shown lethal defects in preimplantation embryo A third KO study showed that Fbxo2 regulates development http://mcb.asm.org/content/26/14/5373.full. amyloid precursor protein levels and processing http://www.jbc.org/content/289/10/7038.long#fn- Rbx1/Roc1: Rbx1 disruption results in early embryonic 1. lethality due to proliferation failure http://www.pnas.org/content/106/15/6203.full.pdf & Fbxo2 has been found to be involved in http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2732615/. neurons, but it has not been tested for potential decreased fertilization or pregnancy rates. It Skp1a: In vivo interference with Skp1 function leads to shows high expression levels in testis, indicating genetic instability and neoplastic transformation potential other not investigated functions http://www.ncbi.nlm.nih.gov/pubmed/12417738?dopt=Abs http://biogps.org/#goto=genereport&id=230904 tract. -

Network-Based Prediction of Polygenic Disease Genes Involved in Cell Motility Miriam Bern†, Alexander King†, Derek A
Bern et al. BMC Bioinformatics 2019, 20(Suppl 12):313 https://doi.org/10.1186/s12859-019-2834-1 RESEARCH Open Access Network-based prediction of polygenic disease genes involved in cell motility Miriam Bern†, Alexander King†, Derek A. Applewhite and Anna Ritz* From International Workshop on Computational Network Biology: Modeling, Analysis and Control Washington, D.C., USA. 29 August 2018 Abstract Background: Schizophrenia and autism are examples of polygenic diseases caused by a multitude of genetic variants, many of which are still poorly understood. Recently, both diseases have been associated with disrupted neuron motility and migration patterns, suggesting that aberrant cell motility is a phenotype for these neurological diseases. Results: We formulate the POLYGENIC DISEASE PHENOTYPE Problem which seeks to identify candidate disease genes that may be associated with a phenotype such as cell motility. We present a machine learning approach to solve this problem for schizophrenia and autism genes within a brain-specific functional interaction network. Our method outperforms peer semi-supervised learning approaches, achieving better cross-validation accuracy across different sets of gold-standard positives. We identify top candidates for both schizophrenia and autism, and select six genes labeled as schizophrenia positives that are predicted to be associated with cell motility for follow-up experiments. Conclusions: Candidate genes predicted by our method suggest testable hypotheses about these genes’ role in cell motility regulation, offering a framework for generating predictions for experimental validation. Keywords: Semi-supervised learning, Functional interaction network, Schizophrenia, Autism, Cell motility Background participants [4, 5], indicating that schizophrenia arises Many polygenic diseases, which arise from the action from a multitude of cellular perturbations compounding or influence of multiple genes, are difficult to geneti- their effects. -

Research Article a Robust Topology-Based Algorithm for Gene Expression Profiling
International Scholarly Research Network ISRN Bioinformatics Volume 2012, Article ID 381023, 11 pages doi:10.5402/2012/381023 Research Article A Robust Topology-Based Algorithm for Gene Expression Profiling Lars Seemann,1 Jason Shulman,2 and Gemunu H. Gunaratne1 1 Department of Physics, University of Houston, Houston, TX 77204, USA 2 Department of Physics, Richard Stockton College of New Jersey, Pomona, NJ 08240, USA Correspondence should be addressed to Gemunu H. Gunaratne, [email protected] Received 3 August 2012; Accepted 30 August 2012 Academic Editors: B. Haubold and K. Yura Copyright © 2012 Lars Seemann et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Early and accurate diagnoses of cancer can significantly improve the design of personalized therapy and enhance the success of therapeutic interventions. Histopathological approaches, which rely on microscopic examinations of malignant tissue, are not conducive to timely diagnoses. High throughput genomics offers a possible new classification of cancer subtypes. Unfortunately, most clustering algorithms have not been proven sufficiently robust. We propose a novel approach that relies on the use of statistical invariants and persistent homology, one of the most exciting recent developments in topology. It identifies a sufficient but compact set of genes for the analysis as well as a core group of tightly correlated patient samples for each subtype. Partitioning occurs hierarchically and allows for the identification of genetically similar subtypes. We analyzed the gene expression profiles of 202 tumors of the brain cancer glioblastoma multiforme (GBM) given at the Cancer Genome Atlas (TCGA) site. -

In This Table Protein Name, Uniprot Code, Gene Name P-Value
Supplementary Table S1: In this table protein name, uniprot code, gene name p-value and Fold change (FC) for each comparison are shown, for 299 of the 301 significantly regulated proteins found in both comparisons (p-value<0.01, fold change (FC) >+/-0.37) ALS versus control and FTLD-U versus control. Two uncharacterized proteins have been excluded from this list Protein name Uniprot Gene name p value FC FTLD-U p value FC ALS FTLD-U ALS Cytochrome b-c1 complex P14927 UQCRB 1.534E-03 -1.591E+00 6.005E-04 -1.639E+00 subunit 7 NADH dehydrogenase O95182 NDUFA7 4.127E-04 -9.471E-01 3.467E-05 -1.643E+00 [ubiquinone] 1 alpha subcomplex subunit 7 NADH dehydrogenase O43678 NDUFA2 3.230E-04 -9.145E-01 2.113E-04 -1.450E+00 [ubiquinone] 1 alpha subcomplex subunit 2 NADH dehydrogenase O43920 NDUFS5 1.769E-04 -8.829E-01 3.235E-05 -1.007E+00 [ubiquinone] iron-sulfur protein 5 ARF GTPase-activating A0A0C4DGN6 GIT1 1.306E-03 -8.810E-01 1.115E-03 -7.228E-01 protein GIT1 Methylglutaconyl-CoA Q13825 AUH 6.097E-04 -7.666E-01 5.619E-06 -1.178E+00 hydratase, mitochondrial ADP/ATP translocase 1 P12235 SLC25A4 6.068E-03 -6.095E-01 3.595E-04 -1.011E+00 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 MIC J3QTA6 CHCHD6 1.090E-04 -5.913E-01 2.124E-03 -5.948E-01 Protein kinase C and casein Q9BY11 PACSIN1 3.837E-03 -5.863E-01 3.680E-06 -1.824E+00 kinase substrate in neurons protein 1 Tubulin polymerization- O94811 TPPP 6.466E-03 -5.755E-01 6.943E-06 -1.169E+00 promoting protein MIC C9JRZ6 CHCHD3 2.912E-02 -6.187E-01 2.195E-03 -9.781E-01 Mitochondrial 2- -
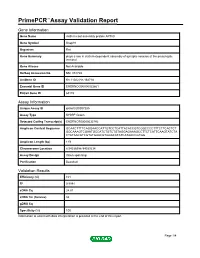
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name clathrin coat assembly protein AP180 Gene Symbol Snap91 Organism Rat Gene Summary plays a role in clathrin-dependent assembly of synaptic vesicles at the presynaptic terminal Gene Aliases Not Available RefSeq Accession No. NM_031728 UniGene ID Rn.11022,Rn.164718 Ensembl Gene ID ENSRNOG00000023861 Entrez Gene ID 65178 Assay Information Unique Assay ID qRnoCID0007285 Assay Type SYBR® Green Detected Coding Transcript(s) ENSRNOT00000032792 Amplicon Context Sequence GCAACTTTTCAGGAACCATTGTCCTCATTACACCGTCGGCCCCTTTCTTCACTCT GGCAAAGTCGAATGCCATCTGTCTGTAGGAGAAAGCCTTCTCATTCAAGTATCTA CTATAACGTCGTATGAACGTGGACATATCATAACCGTGG Amplicon Length (bp) 119 Chromosome Location 8:94036996-94039234 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 101 R2 0.9991 cDNA Cq 24.81 cDNA Tm (Celsius) 82 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Snap91, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector J Mol Neurosci (2013) 50:33–57 DOI 10.1007/s12031-012-9850-1 Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds Pawel Lisowski & Marek Wieczorek & Joanna Goscik & Grzegorz R. Juszczak & Adrian M. Stankiewicz & Lech Zwierzchowski & Artur H. Swiergiel Received: 14 May 2012 /Accepted: 25 June 2012 /Published online: 27 July 2012 # The Author(s) 2012. This article is published with open access at Springerlink.com Abstract There is increasing evidence that depression signaling pathway (Clic6, Drd1a,andPpp1r1b). LA derives from the impact of environmental pressure on transcriptome affected by CMS was associated with genetically susceptible individuals. We analyzed the genes involved in behavioral response to stimulus effects of chronic mild stress (CMS) on prefrontal cor- (Fcer1g, Rasd2, S100a8, S100a9, Crhr1, Grm5,and tex transcriptome of two strains of mice bred for high Prkcc), immune effector processes (Fcer1g, Mpo,and (HA)and low (LA) swim stress-induced analgesia that Igh-VJ558), diacylglycerol binding (Rasgrp1, Dgke, differ in basal transcriptomic profiles and depression- Dgkg,andPrkcc), and long-term depression (Crhr1, like behaviors. We found that CMS affected 96 and 92 Grm5,andPrkcc) and/or coding elements of dendrites genes in HA and LA mice, respectively. Among genes (Crmp1, Cntnap4,andPrkcc) and myelin proteins with the same expression pattern in both strains after (Gpm6a, Mal,andMog). The results indicate significant CMS, we observed robust upregulation of Ttr gene contribution of genetic background to differences in coding transthyretin involved in amyloidosis, seizures, stress response gene expression in the mouse prefrontal stroke-like episodes, or dementia. -

Mapping of SUMO Sites and Analysis of Sumoylation Changes Induced by External Stimuli
Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli Francis Impensa,b,c, Lilliana Radoshevicha,b,c, Pascale Cossarta,b,c,1, and David Ribeta,b,c,1 aUnité des Interactions Bactéries-Cellules, Institut Pasteur, F-75015 Paris, France; bInstitut National de la Santé et de la Recherche Médicale, Unité 604, F-75015 Paris, France; and cInstitut National de la Recherche Agronomique, Unité sous-contrat 2020, F-75015 Paris, France Contributed by Pascale Cossart, July 22, 2014 (sent for review May 28, 2014) SUMOylation is an essential ubiquitin-like modification involved in Mapping the exact lysine residue to which SUMO is attached in important biological processes in eukaryotic cells. Identification of modified proteins is a critical step to get further insight into the small ubiquitin-related modifier (SUMO)-conjugatedresiduesinpro- function of SUMOylation. Indeed, the identification of SUMO teins is critical for understanding the role of SUMOylation but remains sites allows the generation of non-SUMOylatable mutants and the experimentally challenging. We have set up a powerful and high- study of associated phenotypes. Identification of SUMO sites by throughput method combining quantitative proteomics and peptide MS is not straightforward (8). Unlike ubiquitin, which leaves immunocapture to map SUMOylation sites and have analyzed changes a small diglycine (GG) signature tag on the modified lysine resi- in SUMOylation in response to stimuli. With this technique we iden- due after trypsin digestion, SUMO leaves -

Drosophila Tau Negatively Regulates Translation and Olfactory Long-Term Memory, but Facilitates Footshock Habituation and Cytoskeletal Homeostasis
The Journal of Neuroscience, October 16, 2019 • 39(42):8315–8329 • 8315 Cellular/Molecular Drosophila Tau Negatively Regulates Translation and Olfactory Long-Term Memory, But Facilitates Footshock Habituation and Cytoskeletal Homeostasis X Katerina Papanikolopoulou,1* XIlianna G. Roussou,1* XJean Y. Gouzi,1* Martina Samiotaki,2 George Panayotou,2 X Luca Turin,1 and XEfthimios M.C. Skoulakis1 1Institute for Fundamental Biomedical Research, and 2Institute for Bioinnovation, Biomedical Sciences Research Centre “Alexander Fleming,” 16672 Vari, Greece Although the involvement of pathological tau in neurodegenerative dementias is indisputable, its physiological roles have remained elusive in part because its abrogation has been reported without overt phenotypes in mice and Drosophila. This was addressed using the recently described Drosophila tauKO and Mi{MIC} mutants and focused on molecular and behavioral analyses. Initially, we show that Drosophila tau (dTau) loss precipitates dynamic cytoskeletal changes in the adult Drosophila CNS and translation upregulation. Signif- icantly, we demonstrate for the first time distinct roles for dTau in adult mushroom body (MB)-dependent neuroplasticity as its down- regulation within ␣ЈЈneurons impairs habituation. In accord with its negative regulation of translation, dTau loss specifically enhances protein synthesis-dependent long-term memory (PSD-LTM), but not anesthesia-resistant memory. In contrast, elevation of the protein in the MBs yielded premature habituation and depressed PSD-LTM. Therefore, tau loss in Drosophila dynamically alters brain cytoskel- etal dynamics and profoundly affects neuronal proteostasis and plasticity. Key words: Drosophila; habituation; memory; proteome; tau Significance Statement We demonstrate that despite modest sequence divergence, the Drosophila tau (dTau) is a true vertebrate tau ortholog as it interacts with the neuronal microtubule and actin cytoskeleton. -

Potential Sperm Contributions to the Murine Zygote Predicted by in Silico Analysis
REPRODUCTIONRESEARCH Potential sperm contributions to the murine zygote predicted by in silico analysis Panagiotis Ntostis1,2, Deborah Carter3, David Iles1, John Huntriss1, Maria Tzetis2 and David Miller1 1Leeds Institute of Cardiovascular and Metabolic Medicine, University of Leeds, Leeds, West Yorkshire, UK, 2Department of Medical Genetics, St. Sophia’s Children Hospital, School of Medicine, National and Kapodistrian University of Athens, Athens, Attiki, Greece and 3Leeds Institute of Molecular Medicine, University of Leeds, Leeds, West Yorkshire, UK Correspondence should be addressed to D Miller; Email: [email protected] Abstract Paternal contributions to the zygote are thought to extend beyond delivery of the genome and paternal RNAs have been linked to epigenetic transgenerational inheritance in different species. In addition, sperm–egg fusion activates several downstream processes that contribute to zygote formation, including PLC zeta-mediated egg activation and maternal RNA clearance. Since a third of the preimplantation developmental period in the mouse occurs prior to the first cleavage stage, there is ample time for paternal RNAs or their encoded proteins potentially to interact and participate in early zygotic activities. To investigate this possibility, a bespoke next-generation RNA sequencing pipeline was employed for the first time to characterise and compare transcripts obtained from isolated murine sperm, MII eggs and pre-cleavage stage zygotes. Gene network analysis was then employed to identify potential interactions between paternally and maternally derived factors during the murine egg-to-zygote transition involving RNA clearance, protein clearance and post-transcriptional regulation of gene expression. Our in silico approach looked for factors in sperm, eggs and zygotes that could potentially interact co-operatively and synergisticallyp during zygote formation.