Testing the Role of the Red Queen and Court Jester As Drivers of the Macroevolution of Apollo Butterflies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Checklist of Fish and Invertebrates Listed in the CITES Appendices
JOINTS NATURE \=^ CONSERVATION COMMITTEE Checklist of fish and mvertebrates Usted in the CITES appendices JNCC REPORT (SSN0963-«OStl JOINT NATURE CONSERVATION COMMITTEE Report distribution Report Number: No. 238 Contract Number/JNCC project number: F7 1-12-332 Date received: 9 June 1995 Report tide: Checklist of fish and invertebrates listed in the CITES appendices Contract tide: Revised Checklists of CITES species database Contractor: World Conservation Monitoring Centre 219 Huntingdon Road, Cambridge, CB3 ODL Comments: A further fish and invertebrate edition in the Checklist series begun by NCC in 1979, revised and brought up to date with current CITES listings Restrictions: Distribution: JNCC report collection 2 copies Nature Conservancy Council for England, HQ, Library 1 copy Scottish Natural Heritage, HQ, Library 1 copy Countryside Council for Wales, HQ, Library 1 copy A T Smail, Copyright Libraries Agent, 100 Euston Road, London, NWl 2HQ 5 copies British Library, Legal Deposit Office, Boston Spa, Wetherby, West Yorkshire, LS23 7BQ 1 copy Chadwick-Healey Ltd, Cambridge Place, Cambridge, CB2 INR 1 copy BIOSIS UK, Garforth House, 54 Michlegate, York, YOl ILF 1 copy CITES Management and Scientific Authorities of EC Member States total 30 copies CITES Authorities, UK Dependencies total 13 copies CITES Secretariat 5 copies CITES Animals Committee chairman 1 copy European Commission DG Xl/D/2 1 copy World Conservation Monitoring Centre 20 copies TRAFFIC International 5 copies Animal Quarantine Station, Heathrow 1 copy Department of the Environment (GWD) 5 copies Foreign & Commonwealth Office (ESED) 1 copy HM Customs & Excise 3 copies M Bradley Taylor (ACPO) 1 copy ^\(\\ Joint Nature Conservation Committee Report No. -
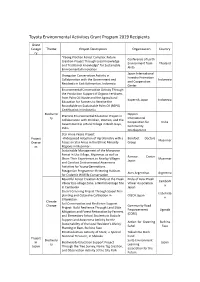
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

The Age of Butterflies Revisited (And Tested) 799
Copyedited by: TE MANUSCRIPT CATEGORY: Systematic Biology Syst. Biol. 68(5):797–813, 2019 © The Author(s) 2019. Published by Oxford University Press on behalf of the Society of Systematic Biologists. This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited. For commercial re-use, please [email protected] DOI:10.1093/sysbio/syz002 Advance Access publication February 25, 2019 Priors and Posteriors in Bayesian Timing of Divergence Analyses: The Age of Butterflies Revisited , , ,∗ NICOLAS CHAZOT1 2 3 ,NIKLAS WAHLBERG1,ANDRÉ VICTOR LUCCI FREITAS4,CHARLES MITTER5,CONRAD , , , LABANDEIRA5 6 7 8,JAE-CHEON SOHN9,RANJIT KUMAR SAHOO10,NOEMY SERAPHIM11,RIENK DE JONG12, AND MARIA HEIKKILÄ13 1Department of Biology, Lunds Universitet, Sölvegatan 37, 223 62 Lund, Sweden; 2Gothenburg Global Biodiversity Centre, Box 461, 405 30 Gothenburg, Sweden; 3Department of Biological and Environmental Sciences, University of Gothenburg, Box 461, 405 30 Gothenburg, Sweden; 4Departamento de Biologia Animal, Instituto de Biologia, Universidade Estadual de Campinas (UNICAMP), Cidade Universitária Zeferino Vaz, Caixa Postal 6109, Barão Geraldo 13083-970, Campinas, São Paulo, Brazil; 5Department of Entomology, University of Maryland, 4291 Fieldhouse Dr, College Park, MD 20742, USA; 6Department of Paleobiology, National -

Spotting Butterflies Says Ulsh
New & Features “A butterfly’s lifespan generally cor- responds with the size of the butterfly,” Spotting Butterflies says Ulsh. The tiny blues often seen in the mountains generally only live about How, when and where to find Lepidoptera in 10 days. Some species, however, will the Cascades and Olympics overwinter in the egg, pupa or chrysalid form (in the cocoon prior to becoming winged adults). A few Northwest spe- cies overwinter as adults, and one—the mourning cloak—lives for ten months, and is the longest lived butterfly in North America. The first thing that butterflies do upon emerging from the chrysalis and unfold- ing their wings is to breed. In their search for mates, some butterflies “hilltop,” or stake out spots on high trees or ridgelines to make themselves more prominent. A butterfly’s wing colors serve two distinct purposes. The dorsal, or upperside of the wings, are colorful, and serve to attract mates. The ventral, or underside of the wings generally serves to camouflage the insects. So a butterfly such as the satyr comma has brilliant orange and yellow Western tiger and pale tiger swallowtail butterflies “puddling.” When looking for for spots when seen with wings open, and butterflies along the trail keep an eye on moist areas or meadows with flowers. a bark-like texture to confuse predators when its wings are closed. By Andrew Engelson Butterfly Association, about where, how As adults, butterflies also seek out Photos by Idie Ulsh and when to look for butterflies in our nectar and water. Butterflies generally mountains. Ulsh is an accomplished Butterflies are the teasers of wildlife. -

Testing the Role of the Red Queen and Court Jester As Drivers of The
bioRxiv preprint doi: https://doi.org/10.1101/198960; this version posted October 5, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Running head 2 TESTING THE RED QUEEN AND COURT JESTER 3 4 Title 5 Testing the role of the Red Queen and Court Jester as drivers of the 6 macroevolution of Apollo butterflies 7 8 Authors 1,2,3 4 5 9 FABIEN L. CONDAMINE *, JONATHAN ROLLAND , SEBASTIAN HÖHNA , FELIX A. H. 3 2 10 SPERLING † AND ISABEL SANMARTÍN † 11 12 Authors’ addresses 13 1 CNRS, UMR 5554 Institut des Sciences de l’Evolution (Université de Montpellier | CNRS 14 | IRD | EPHE), Montpellier, France; 15 2 Department of Biodiversity and Conservation, Real Jardín Botánico, CSIC, Plaza de 16 Murillo, 2; 28014 Madrid, Spain; 17 3 Department of Biological Sciences, University of Alberta, Edmonton T6G 2E9, AB, Canada; 18 4 Department of Ecology and Evolution, University of Lausanne, 1015 Lausanne, Switzerland; 19 5 Division of Evolutionary Biology, Ludwig-Maximilian-Universität München, Grosshaderner 20 Strasse 2, Planegg-Martinsried 82152, Germany 21 22 † Co-senior authors. 23 Corresponding author (*): Fabien L. Condamine, CNRS, UMR 5554 Institut des Sciences de 24 l’Evolution (Université de Montpellier), Place Eugène Bataillon, 34095 Montpellier, France. 25 Phone: +336 749 322 96 | E-mail: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/198960; this version posted October 5, 2017. -

XII Dr. S. Pradhan Memorial Lecture Entomofauna, Ecosystem And
XII Dr. S. Pradhan Memorial Lecture September 28, 2020 Entomofauna, Ecosystem and Economics by Dr. Kailash Chandra, Director, Zoological Survey of India, Kolkata DIVISION OF ENTOMOLOGY, ICAR-INDIAN AGRICULTURAL RESEARCH INSTITUTE, NEW DELHI- 110012 ORGANIZING COMMITTEE PATRON Dr. A. K. Singh, Director, ICAR-IARI, New Delhi CONVENER Dr. Debjani Dey, Head (Actg.), Division of Entomology MEMBERS Dr. H. R. Sardana, Director, ICAR-NCIPM, New Delhi Dr. Subhash Chander, Professor & Principal Scientist Dr. Bishwajeet Paul, Principal Scientist Dr. Naresh M. Meshram, Senior Scientist Mrs. Rajna S, Scientist Dr. Bhagyasree S N, Scientist Dr. S R Sinha, CTO Shri Sushil Kumar, AAO (Member Secretary) XIII Dr. S. Pradhan Memorial Lecture September 28, 2020 Entomofauna, Ecosystem and Economics by Dr. Kailash Chandra, Director, Zoological Survey of India, Kolkata DIVISION OF ENTOMOLOGY, ICAR-INDIAN AGRICULTURAL RESEARCH INSTITUTE, NEW DELHI- 110012 Dr. S. Pradhan May 13, 1913 - February 6, 1973 4 Dr. S. Pradhan - A Profile Dr. S. Pradhan, a doyen among entomologists, during his 33 years of professional career made such an impact on entomological research and teaching that Entomology and Plant Protection Science in India came to the forefront of agricultural research. His success story would continue to enthuse Plant Protection Scientists of the country for generations to come. The Beginning Shyam Sunder Lal Pradhan had a humble beginning. He was born on May 13, 1913, at village Dihwa in Bahraich district of Uttar Pradesh. He came from a middle class family. His father, Shri Gur Prasad Pradhan, was a village level officer of the state Government having five sons and three daughters. -

Amphiesmeno- Ptera: the Caddisflies and Lepidoptera
CY501-C13[548-606].qxd 2/16/05 12:17 AM Page 548 quark11 27B:CY501:Chapters:Chapter-13: 13Amphiesmeno-Amphiesmenoptera: The ptera:Caddisflies The and Lepidoptera With very few exceptions the life histories of the orders Tri- from Old English traveling cadice men, who pinned bits of choptera (caddisflies)Caddisflies and Lepidoptera (moths and butter- cloth to their and coats to advertise their fabrics. A few species flies) are extremely different; the former have aquatic larvae, actually have terrestrial larvae, but even these are relegated to and the latter nearly always have terrestrial, plant-feeding wet leaf litter, so many defining features of the order concern caterpillars. Nonetheless, the close relationship of these two larval adaptations for an almost wholly aquatic lifestyle (Wig- orders hasLepidoptera essentially never been disputed and is supported gins, 1977, 1996). For example, larvae are apneustic (without by strong morphological (Kristensen, 1975, 1991), molecular spiracles) and respire through a thin, permeable cuticle, (Wheeler et al., 2001; Whiting, 2002), and paleontological evi- some of which have filamentous abdominal gills that are sim- dence. Synapomorphies linking these two orders include het- ple or intricately branched (Figure 13.3). Antennae and the erogametic females; a pair of glands on sternite V (found in tentorium of larvae are reduced, though functional signifi- Trichoptera and in basal moths); dense, long setae on the cance of these features is unknown. Larvae do not have pro- wing membrane (which are modified into scales in Lepi- legs on most abdominal segments, save for a pair of anal pro- doptera); forewing with the anal veins looping up to form a legs that have sclerotized hooks for anchoring the larva in its double “Y” configuration; larva with a fused hypopharynx case. -

Anthropogenic Threats to High-Altitude Parnassian Diversity Fabien Condamine, Felix Sperling
Anthropogenic threats to high-altitude parnassian diversity Fabien Condamine, Felix Sperling To cite this version: Fabien Condamine, Felix Sperling. Anthropogenic threats to high-altitude parnassian diversity. News of The Lepidopterists’ Society, 2018, 60, pp.94-99. hal-02323624 HAL Id: hal-02323624 https://hal.archives-ouvertes.fr/hal-02323624 Submitted on 23 Oct 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. _______________________________________________________________________________________News of The Lepidopterists’ Society Volume 60, Number 2 Conservation Matters: Contributions from the Conservation Committee Anthropogenic threats to high-altitude parnassian diversity Fabien L. Condamine1 and Felix A.H. Sperling2 1CNRS, UMR 5554 Institut des Sciences de l’Evolution (Université de Montpellier), Place Eugène Bataillon, 34095 Montpellier, France [email protected], corresponding author 2Department of Biological Sciences, University of Alberta, Edmonton T6G 2E9, Alberta, Canada Introduction extents (Chen et al. 2011). However, this did not result in increases in range area because the area of land available Global mean annual temperatures increased by ~0.85° declines with increasing elevation. Accordingly, extinc- C between 1880 and 2012 and are likely to rise by an tion risk may increase long before species reach a summit, additional 1° C to 4° C by 2100 (Stocker et al. -

Whole Genome Shotgun Phylogenomics Resolves the Pattern
Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution Rémi Allio, Celine Scornavacca, Benoit Nabholz, Anne-Laure Clamens, Felix Sperling, Fabien Condamine To cite this version: Rémi Allio, Celine Scornavacca, Benoit Nabholz, Anne-Laure Clamens, Felix Sperling, et al.. Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution. Systematic Biology, Oxford University Press (OUP), 2020, 69 (1), pp.38-60. 10.1093/sysbio/syz030. hal-02125214 HAL Id: hal-02125214 https://hal.archives-ouvertes.fr/hal-02125214 Submitted on 10 May 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Running head Shotgun phylogenomics and molecular dating Title proposal Downloaded from https://academic.oup.com/sysbio/advance-article-abstract/doi/10.1093/sysbio/syz030/5486398 by guest on 07 May 2019 Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution Authors Rémi Allio1*, Céline Scornavacca1,2, Benoit Nabholz1, Anne-Laure Clamens3,4, Felix -

Biodiversity and Ecosystem Management in the Iraqi Marshlands
Biodiversity and Ecosystem Management in the Iraqi Marshlands Screening Study on Potential World Heritage Nomination Tobias Garstecki and Zuhair Amr IUCN REGIONAL OFFICE FOR WEST ASIA 1 The designation of geographical entities in this book, and the presentation of the material, do not imply the expression of any opinion whatsoever on the part of IUCN concerning the legal status of any country, territory, or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The views expressed in this publication do not necessarily reflect those of IUCN. Published by: IUCN ROWA, Jordan Copyright: © 2011 International Union for Conservation of Nature and Natural Resources Reproduction of this publication for educational or other non-commercial purposes is authorized without prior written permission from the copyright holder provided the source is fully acknowledged. Reproduction of this publication for resale or other commercial purposes is prohibited without prior written permission of the copyright holder. Citation: Garstecki, T. and Amr Z. (2011). Biodiversity and Ecosystem Management in the Iraqi Marshlands – Screening Study on Potential World Heritage Nomination. Amman, Jordan: IUCN. ISBN: 978-2-8317-1353-3 Design by: Tobias Garstecki Available from: IUCN, International Union for Conservation of Nature Regional Office for West Asia (ROWA) Um Uthaina, Tohama Str. No. 6 P.O. Box 942230 Amman 11194 Jordan Tel +962 6 5546912/3/4 Fax +962 6 5546915 [email protected] www.iucn.org/westasia 2 Table of Contents 1 Executive -

Tera: Papilionidae): Cladistic Reappraisals Using Mainly Immature Stage Characters, with Focus on the Birdwings Ornithoptera Boisduval
Bull. Kitakyushu Mus. Nat. Hist., 15: 43-118. March 28, 1996 Gondwanan Evolution of the Troidine Swallowtails (Lepidop- tera: Papilionidae): Cladistic Reappraisals Using Mainly Immature Stage Characters, with Focus on the Birdwings Ornithoptera Boisduval Michael J. Parsons Entomology Section, Natural History Museum of Los Angeles County 900 Exposition Blvd., LosAngeles, California 90007, U.S.A.*' (Received December 13, 1995) Abstract In order to reappraise the interrelationships of genera in the tribe Troidini, and to test the resultant theory of troidine evolution against biogeographical data a cladistic analysis of troidine genera was performed. Data were obtained mainly from immature stages, providing characters that appeared to be more reliable than many "traditional" adult characters. A single cladogram hypothesising phylogenetic relation ships of the troidine genera was generated. This differs markedly from cladograms obtained in previous studies that used only adult characters. However, the cladogram appears to fit well biogeographical data for the Troidini in terms of vicariance biogcography, especially as this relates to the general hypotheses of Gondwanaland fragmentation and continental drift events advanced by recent geological studies. The genus Ornithoptera is shown to be distinct from Troides. Based on input data drawn equally from immature stages and adult characters, a single cladogram hypothesising the likely phylogeny of Ornithoptera species was generated. With minor weighting of a single important adult character (male -

Phylogeography of Koramius Charltonius (Gray, 1853) (Lepidoptera: Papilionidae): a Case of Too Many Poorly Circumscribed Subspecies
©Societas Europaea Lepidopterologica; download unter http://www.soceurlep.eu/ und www.zobodat.at Nota Lepi. 39(2) 2016: 169–191 | DOI 10.3897/nl.39.7682 Phylogeography of Koramius charltonius (Gray, 1853) (Lepidoptera: Papilionidae): a case of too many poorly circumscribed subspecies Stanislav K. Korb1, Zdenek F. Fric2, Alena Bartonova2 1 Institute of Biology, Komi Science Center, Ural Division of the Russian Academy of Sciences, Kommunisticheskaya 28, 167982 Syktyvkar, Russia; [email protected] 2 Institute of Entomology, Biology Centre, Academy of Sciences of the Czech Republic, Branisovska 31, 370 05 Ceske Budejovice, Czech Republic; [email protected] http://zoobank.org/670C4AB9-A810-4F7E-A3FC-B433940CFD67 Received 2 January 2016; accepted 3 October 2016; published: 27 October 2016 Subject Editor: Thomas Schmitt. Abstract. Koramius charltonius (Gray, 1853) (Lepidoptera: Papilionidae) is distributed in the mountains of Central Asia. We analysed genetic and phylogeographic patterns throughout the western part of its range using a mitochondrial marker (COI). We also analysed the wing pattern using multivariate statistics. We found that the species contains several unique haplotypes in the west and shared haplotypes in the east. The haplotype groups do not correspond to the wing pattern and also the described subspecies do not correspond to either the haplotypes or the groups circumscribed by the wing pattern. Currently, there are more than ten subspecies of K. charltonius in Central Asia; based on our analyses we suggest a reduction to only five of them. The fol- lowing nomenclatural changes are applied: (1) K. charltonius aenigma Dubatolov & Milko, 2003, syn. n., K. charltonius sochivkoi Churkin, 2009, syn.n., and K.