Step I: White Paper Application
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Abstract Pultorak, Elizabeth Lauren
ABSTRACT PULTORAK, ELIZABETH LAUREN. The Epidemiology of Lyme Disease and Bartonellosis in Humans and Animals. (Under the direction of Edward B. Breitschwerdt). The expansion of vector borne diseases in humans, a variety of mammalian hosts, and arthropod vectors draws attention to the need for enhanced diagnostic techniques for documenting infection in hosts, effective vector control, and treatment of individuals with associated diseases. Through improved diagnosis of vector-borne disease in both humans and animals, epidemiological studies to elucidate clinical associations or spatio-temporal relationships can be assessed. Veterinarians, through the use of the C6 peptide in the SNAP DX test kit, may be able to evaluate the changing epidemiology of borreliosis through their canine population. We developed a survey to evaluate the practices and perceptions of veterinarians in North Carolina regarding borreliosis in dogs across different geographic regions of the state. We found that veterinarians’ perception of the risk of borreliosis in North Carolina was consistent with recent scientific reports pertaining to geographic expansion of borreliosis in the state. Veterinarians should promote routine screening of dogs for Borrelia burgdorferi exposure as a simple, inexpensive form of surveillance in this transitional geographic region. We next conducted two separate studies to evaluate Bartonella spp. bacteremia or presence of antibodies against B. henselae, B. koehlerae, or B. vinsonii subsp. berkhoffii in 296 patients examined by a rheumatologist and 192 patients with animal exposure (100%) and recent animal bites and scratches (88.0%). Among 296 patients examined by a rheumatologist, prevalence of antibodies (185 [62%]) and Bartonella spp. bacteremia (122 [41.1%]) was high. -

Beating Chronic LYME
Section 1 Beating Chronic LYME Dr. Kevin Conners Fellowship in Integrative Cancer Therapy Fellowship in Anti-Aging, Regenerative, and Functional Medicine American Academy of Anti-Aging Medicine www.ConnersClinic.com From left to right: Larvae, Nymph, Female, Male Tick Tick in Nymph stage is the size of a poppy seed. Beating Chronic LYME Forward I used to live in the woods. My wife and four children at the time purchased 280 acres in Wisconsin and basically lived off the land. We grew most of our own food, were ‘off grid’ as we produced our own electricity through solar panels, and had to pump our water by hand. It was certainly a different way of life that prepared us for missionary work in Mexico. It was 1997 and at the time I had begun hearing about Lyme disease being a tick- born disorder. I had never seen a deer tick before moving to our ‘little house in the big woods’ but one thing was for certain – we had plenty of deer. They were as populous as the mosquitoes. To make a long story short, both my oldest daughter and I had contracted Lyme during our three year stay. I experienced the violent sickness of acute Lyme as well as a beautiful bulls-eye rash that made the diagnosis easy. I took just 3 days of antibiotics and since that was just the second time that I had ever taken a prescription medication in my life, they eradicated the disease effectively. My daughter on the other hand wasn’t so fortunate. She never had an acute illness and we never saw any rash therefore we didn’t catch the disease until it had advanced to Chronic Lyme Disease (CLD). -

Detection and Partial Molecular Characterization of Rickettsia and Bartonella from Southern African Bat Species
Detection and partial molecular characterization of Rickettsia and Bartonella from southern African bat species by Tjale Mabotse Augustine (29685690) Submitted in partial fulfillment of the requirements for the degree MAGISTER SCIENTIAE (MICROBIOLOGY) in the Department of Microbiology and Plant Pathology Faculty of Natural and Agricultural Sciences University of Pretoria Pretoria, South Africa Supervisor: Dr Wanda Markotter Co-supervisors: Prof Louis H. Nel Dr Jacqueline Weyer May, 2012 I declare that the thesis, which I hereby submit for the degree MSc (Microbiology) at the University of Pretoria, South Africa, is my own work and has not been submitted by me for a degree at another university ________________________________ Tjale Mabotse Augustine i Acknowledgements I would like send my sincere gratitude to the following people: Dr Wanda Markotter (University of Pretoria), Dr Jacqueline Weyer (National Institute for Communicable Diseases-National Health Laboratory Service) and Prof Louis H Nel (University of Pretoria) for their supervision and guidance during the project. Dr Jacqueline Weyer (Centre for Zoonotic and Emerging diseases (Previously Special Pathogens Unit), National Institute for Communicable Diseases (National Heath Laboratory Service), for providing the positive control DNA for Rickettsia and Dr Jenny Rossouw (Special Bacterial Pathogens Reference Unit, National Institute for Communicable Diseases-National Health Laboratory Service), for providing the positive control DNA for Bartonella. Dr Teresa Kearney (Ditsong Museum of Natural Science), Gauteng and Northern Region Bat Interest Group, Kwa-Zulu Natal Bat Interest Group, Prof Ara Monadjem (University of Swaziland), Werner Marias (University of Johannesburg), Dr Francois du Rand (University of Johannesburg) and Prof David Jacobs (University of Cape Town) for collection of blood samples. -

Bartonella: Emerging Pathogen Or Emerging Awareness?
International Journal of Infectious Diseases (2009) 13, 3—8 http://intl.elsevierhealth.com/journals/ijid PERSPECTIVE Bartonella: emerging pathogen or emerging awareness? Elin Mogollon-Pasapera, Laszlo Otvos Jr, Antonio Giordano, Marco Cassone * Sbarro Health Research Organization, College of Science and Technology, Temple University, BioLifeScience Building suite 419, 1900 N 12th Street, 19122 Philadelphia, PA, USA Received 17 October 2007; received in revised form 26 January 2008; accepted 14 April 2008 Corresponding Editor: William Cameron, Ottawa, Canada KEYWORDS Summary The number of known Bartonella species is rapidly growing. Some of them are Bartonella; responsible for distinct infectious diseases and show different prevalence and antibiotic suscept- Carrion’s disease; ibility profiles. Not only have some vectors of Bartonella not been fully characterized, but also Epidemiology; intermediate hosts are actually much more numerous and diverse than previously thought. Among Cat-scratch disease; these, dogs differ from cats because they tend to suffer an overt disease similar to humans, thus Therapy providing the base for a useful animal indicator and research model. Among the debilitating conditions with an unclear impact on the course of these infections, specific conditions (e.g., homelessness, alcoholism) have been linked to a much higher prevalence and to high risk of unfavorable outcome. Due to the limited arsenal of antibiotics effective in vivo on this peculiar intracellular pathogen, the risk/benefit balance of antibiotic therapy is sometimes difficult to draw. In this evolving picture, the recent discoveries of new species highlights the importance of basic molecular biology resources that would bring major public health benefits if available in endemic areas, and specifically in many areas of Peru and Bolivia. -

Characterizing the Molecular Biology of a Bacteriophage-Like Particle from Bartonella Bacilliformis
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 1999 Characterizing the molecular biology of a bacteriophage-like particle from Bartonella bacilliformis Kent D. Barbian The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Barbian, Kent D., "Characterizing the molecular biology of a bacteriophage-like particle from Bartonella bacilliformis" (1999). Graduate Student Theses, Dissertations, & Professional Papers. 6634. https://scholarworks.umt.edu/etd/6634 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. i I S Maureen and Mike MANSFIELD LIBRARY The University of IVIONTANA Permission is granted by the author to reproduce this material in its entirety, provided that this material is used for scholarly purposes and is properly cited in published works and reports. ** Please check ’’Yes'* or ”No” and provide signature ** Yes, I grant permission X" No, I do not grant permission _____ Author's Signature Date Any copying for conunercial purposes or financial gain may be undertaken only with the author's explicit consent. Characterizing the Molecular Biology of a Bacteriophage-Like Particle FromBartonella bacilliformis by Kent D. Barbian B.A,, The University of Montana, 1997 Presented in partial fulfillment of the requirements for the degree of Master of Science in Microbiology The University of Montana 1999 Approved by: Committee Chair Dean, Graduate School Date UMI Number: EP37435 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -
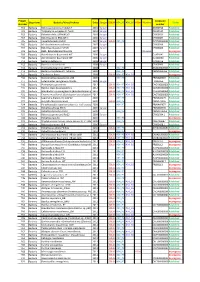
Project Number Organisms Bacteria/Virus/Archaea Date
Project_ Accession Organisms Bacteria/Virus/Archaea Date Sanger SOLiD 454_PE 454_SG PGM Illumina Status Number number P01 Bacteria Rickettsia conorii str.Malish 7 2001 Sanger AE006914 Published P02 Bacteria Tropheryma whipplei str.Twist 2003 Sanger AE014184 Published P03 Bacteria Rickettsia felis URRWXCal2 2005 Sanger CP000053 Published P04 Bacteria Rickettsia bellii RML369-C 2006 Sanger CP000087 Published P05 Bacteria Coxiella burnetii CB109 2007 Sanger SOLiD 454_PE AKYP00000000 Published P06 Bacteria Minibacterium massiliensis 2007 Sanger CP000269 Published P07 Bacteria Rickettsia massiliae MTU5 2007 Sanger CP000683 Published P08 Bacteria BaBL=Bête à Bernard Lascola 2007 Illumina In progress P09 Bacteria Acinetobacter baumannii AYE 2006 Sanger CU459141 Published P10 Bacteria Acinetobacter baumannii SDF 2006 Sanger CU468230 Published P11 Bacteria Borrelia duttonii Ly 2008 Sanger CP000976 Published P12 Bacteria Borrelia recurrentis A1 2008 Sanger CP000993 Published P13 Bacteria Francisella tularensis URFT1 2008 454_PE ABAZ00000000Published P14 Bacteria Borrelia crocidurae str. Achema 2009 454_PE PRJNA162335 Published P15 Bacteria Citrobacter koseri 2009 SOLiD 454_PE 454_SG In progress P16 Bacteria Diplorickettsia massiliensis 20B 2009 454_PE PRJNA86907 Published P17 Bacteria Enterobacter aerogenes EA1509E 2009 Sanger FO203355 Published P18 Bacteria Actinomyces grossensis 2012 SOLiD 454_PE 454_SG CAGY00000000Published P19 Bacteria Bacillus massiliosenegalensis 2012 SOLiD 454_PE 454_SG CAHJ00000000 Published P20 Bacteria Brevibacterium senegalensis -

Bartonellosis Ed Breitschwerdt, DVM (Ed [email protected]) Tests Available at VBDDL: Serology (IFA) and PCR on Blood Or CSF
Bartonellosis Ed Breitschwerdt, DVM ([email protected]) Tests available at VBDDL: Serology (IFA) and PCR on blood or CSF There are a growing number of Bartonella species associated with disease manifestations in cats, dogs, humans and other less domesticated host species including Bartonella henselae, B. vinsonii subspecies berkhoffii, B.rochalimae, B.clarridgeiae, and B. koehlerae. Genetically unique strains or genotypes are being described within each species. This may affect test interpretation because most laboratories only test for a limited selection of species and serological cross reactivity is not always present. Knowledge of which species are tested for is important for both antibody and DNA testing. Risk factors: o History of flea, tick, biting fly, keds, lice, or sandfly exposure Disease: o Transient lethargy o Fever o Lymphadenopathy o Gingivitis o Stomatitis o Uveitis o Neurologic dysfunction o Endocarditis o Retinal disease Bartonella henselae – B.henselae is a flea-transmitted zoonotic pathogen with the cat identified as a major reservoir for human infection. Fever and bacteremia, endocarditis, lymphadenopathy (cat scratch disease in people), bacillary angiomatosis, neurologic dysfunction and retinal disease can be caused by B. henselae, particularly in immunocompromised individuals. Following flea transmission or blood transfusion to cats, B. henselae causes a relapsing pattern of bacteremia, persisting for months to years. Bartonella koehlerae – B.koehlerae is a zoonotic bacterium that has but rarely been reported as an infectious cause of disease in dogs or human patients. Cats are considered the primary reservoir host for B. koehlerae as with B. henselae. Ctenocephalides felis, the cat flea, is considered a transmission competent vector for both organisms. -

Bartonella Rochalimae and B. Vinsonii Subsp. Berkhoffii in Wild Carnivores from Colorado, Usa
DOI: 10.7589/2016-01-015 Journal of Wildlife Diseases, 52(4), 2016, pp. 844–849 Ó Wildlife Disease Association 2016 BARTONELLA ROCHALIMAE AND B. VINSONII SUBSP. BERKHOFFII IN WILD CARNIVORES FROM COLORADO, USA Ying Bai,1,4 Amy Gilbert,2 Karen Fox,3 Lynn Osikowicz,1 and Michael Kosoy1 1 Bacterial Disease Branch, Division of Vector-Borne Disease, Centers for Disease Control and Prevention, 3156 Rampart Rd., Fort Collins, Colorado 80521, USA 2 National Wildlife Research Center, USDA/APHIS/Wildlife Services, 4101 Laporte Ave., Fort Collins, Colorado 80521, USA 3 Colorado Parks and Wildlife, 317 W Prospect Rd., Fort Collins, Colorado 80525, USA 4 Corresponding author (email: [email protected]) Downloaded from http://meridian.allenpress.com/jwd/article-pdf/52/4/844/2239205/2016-01-015.pdf by guest on 28 September 2021 ABSTRACT: Spleen samples from 292 wild carnivores from Colorado, US were screened for Bartonella infection. Bartonella DNA was detected in coyotes (Canis latrans) (28%), striped skunks (Mephitis mephitis) (23%), red foxes (Vulpes vulpes) (27%), and raccoons (Procyon lotor) (8%) but not in black bears (Ursus americanus), gray foxes (Urocyon cinereoargenteus), and mountain lions (Puma concolor). Two Bartonella species, B. vinsonii subsp. berkhoffii and B. rochalimae, were identified. All 10 infected striped skunks exclusively carried B. rochalimae while coyotes, red foxes, and raccoons could be infected with both Bartonella species. Five of seven infected coyotes carried B. v. berkhoffii whereas five of seven infected red foxes and 11 of 14 infected raccoons carried B. rochalimae. Further studies are needed to understand relationships between Bartonella species, wild carnivores, and their ectoparasites. -

Bartonella Infections in Cats and Dogs Including Zoonotic Aspects Alejandra Álvarez-Fernández1, Edward B
Álvarez-Fernández et al. Parasites & Vectors (2018)11:624 https://doi.org/10.1186/s13071-018-3152-6 REVIEW Open Access Bartonella infections in cats and dogs including zoonotic aspects Alejandra Álvarez-Fernández1, Edward B. Breitschwerdt2 and Laia Solano-Gallego1* Abstract Bartonellosis is a vector-borne zoonotic disease with worldwide distribution that can infect humans and a large number of mammals including small companion animals (cats and dogs). In recent years, an increasing number of studies from around the world have reported Bartonella infections, although publications have predominantly focused on the North American perspective. Currently, clinico-pathological data from Europe are more limited, suggesting that bartonellosis may be an infrequent or underdiagnosed infectious disease in cats and dogs. Research is needed to confirm or exclude Bartonella infection as a cause of a spectrum of feline and canine diseases. Bartonella spp. can cause acute or chronic infections in cats, dogs and humans. On a comparative medical basis, different clinical manifestations, such as periods of intermittent fever, granulomatous inflammation involving the heart, liver, lymph nodes and other tissues, endocarditis, bacillary angiomatosis, peliosis hepatis, uveitis and vasoproliferative tumors have been reported in cats, dogs and humans. The purpose of this review is to provide an update and European perspective on Bartonella infections in cats and dogs, including clinical, diagnostic, epidemiological, pathological, treatment and zoonotic aspects. Keywords: Bartonella, Dog, Cat, Europe, Zoonosis Background diagnostic, epidemiological, pathological, treatment and Bartonella is a genus of Alphaproteobacteria within the zoonotic aspects. family Bartonellaceae. Bartonella spp. are small, thin, short and slightly curved, gram-negative, hemotropic Bartonella and rod-shaped bacteria [1]. -

Ru 2015 150 263 a (51) Мпк A61k 31/155 (2006.01)
РОССИЙСКАЯ ФЕДЕРАЦИЯ (19) (11) (13) RU 2015 150 263 A (51) МПК A61K 31/155 (2006.01) ФЕДЕРАЛЬНАЯ СЛУЖБА ПО ИНТЕЛЛЕКТУАЛЬНОЙ СОБСТВЕННОСТИ (12) ЗАЯВКА НА ИЗОБРЕТЕНИЕ (21)(22) Заявка: 2015150263, 01.05.2014 (71) Заявитель(и): НЕОКУЛИ ПТИ ЛТД (AU) Приоритет(ы): (30) Конвенционный приоритет: (72) Автор(ы): 01.05.2013 AU 2013901517 ПЕЙДЖ Стефен (AU), ГАРГ Санджай (AU) (43) Дата публикации заявки: 06.06.2017 Бюл. № 16 RU (85) Дата начала рассмотрения заявки PCT на национальной фазе: 01.12.2015 (86) Заявка PCT: AU 2014/000480 (01.05.2014) 2015150263 (87) Публикация заявки PCT: WO 2014/176634 (06.11.2014) Адрес для переписки: 190000, Санкт-Петербург, Box-1125, "ПАТЕНТИКА" A (54) СПОСОБЫ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ (57) Формула изобретения 1. Способ лечения или профилактики бактериальной колонизации или инфекции у субъекта, включающий стадию: введения субъекту терапевтически эффективного количества робенидина или его терапевтически приемлемой соли, причем указанная A бактериальная колонизация или инфекция вызвана бактериальным агентом. 2. Способ по п. 1, отличающийся тем, что субъект выбран из группы, включающей: человека, животных, принадлежащих видам семейства псовых, кошачьих, крупного рогатого скота, овец, коз, свиней, птиц, рыб и лошадей. 3. Способ по п. 1, отличающийся тем, что робенидин вводят субъекту в дозе в диапазоне от 0,1 до 250 мг/кг массы тела. 4. Способ по любому из пп. 1-3, отличающийся тем, что бактериальный агент является 2015150263 грамположительным. 5. Способ по п. 4, отличающийся тем, что бактериальный агент выбран из -

Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales
Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales TECHNICAL BIOSAFETY COMMITTEE (TBC) NATIONAL CENTER FOR GENETIC ENGINEERING AND BIOTECHNOLOGY (BIOTEC) NATIONAL SCIENCE AND TECHNOLOGY DEVELOPMENT AGENCY (NSTDA) MINISTRY OF SCIENCE AND TECHNOLOGY (MOST) 2015 Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales TECHNICAL BIOSAFETY COMMITTEE (TBC) NATIONAL CENTER FOR GENETIC ENGINEERING AND BIOTECHNOLOGY (BIOTEC) NATIONAL SCIENCE AND TECHNOLOGY DEVELOPMENT AGENCY (NSTDA) MINISTRY OF SCIENCE AND TECHNOLOGY (MOST) 2015 Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales Technical Biosafety Committee National Center for Genetic Engineering and Biotechnology National Science and Technology Development Agency (NSTDA) © National Center for Genetic Engineering and Biotechnology 2015 ISBN : 978-616-12-0386-3 Tel : +66(0)2-564-6700 Fax : +66(0)2-564-6703 E-mail : [email protected] URL : http://www.biotec.or.th Printing House : P.A. Living Printing Co.,Ltd 4 Soi Sirintron 7 Road Sirintron District Bangplad Province Bangkok 10700 Tel : +66(0)2-881 9890 Fax : +66(0)2-881 9894 Preface Genetically Modified Microorganisms (GMMs) were first used in B.E. 2525 to produce insulin in industrial medicine. Currently, GMMs are used in various industries, such as the food, pharmaceutical and bioplastic industries, to manufacture a number of important consumer products. To ensure operator and environmental safety, the Technical Biosafety Committee (TBC) of the National Center for Genetic Engineering and Biotechnology (BIOTEC), the National Science and Technology Development Agency (NSTDA), has prepared guidelines for GMM work, publishing “Biosafety Guidelines for Contained Use of Genetically Modified Microorganisms at Pilot and Industrial Scales” in B.E. -

Hydropotes Inermis Argyropus)
ISSN (Print) 0023-4001 ISSN (Online) 1738-0006 Korean J Parasitol Vol. 54, No. 1: 87-91, February 2016 ▣ BRIEF COMMUNICATION http://dx.doi.org/10.3347/kjp.2016.54.1.87 Prevalence of Anaplasma and Bartonella spp. in Ticks Collected from Korean Water Deer (Hydropotes inermis argyropus) Jun-Gu Kang1, Sungjin Ko1, Heung-Chul Kim2, Sung-Tae Chong2, Terry A. Klein3, Jeong-Byoung Chae1, 1 4 5 6 7 1, Yong-Sun Jo , Kyoung-Seong Choi , Do-Hyeon Yu , Bae-Keun Park , Jinho Park , Joon-Seok Chae * 1Laboratory of Veterinary Internal Medicine, BK21 PLUS Program for Creative Veterinary Science Research, Research Institute for Veterinary Science and College of Veterinary Medicine, Seoul National University, Seoul 08826, Korea; 25th Medical Detachment, 168th Multifunctional Medical Battalion, 65th Medical Brigade, Unit 15247, APO AP96205-5247, USA; 3Public Health Command District-Korea, 65th Medical Brigade, Unit 15281, APO AP 96205-5281, USA; 4College of Ecology and Environmental Science, Kyungpook National University, Sangju 37224, Korea; 5College of Veterinary Medicine, Chonnam National University, Gwangju 61186, Korea; 6College of Veterinary Medicine, Chungnam National University, Daejeon 34134, Korea; 7College of Veterinary Medicine, Chonbuk National University, Iksan 54596, Korea Abstract: Deer serve as reservoirs of tick-borne pathogens that impact on medical and veterinary health worldwide. In the Republic of Korea, the population of Korean water deer (KWD, Hydropotes inermis argyropus) has greatly increased from 1982 to 2011, in part, as a result of reforestation programs established following the Korean War when much of the land was barren of trees. Eighty seven Haemaphysalis flava, 228 Haemaphysalis longicornis, 8 Ixodes nipponensis, and 40 Ixodes persulcatus (21 larvae, 114 nymphs, and 228 adults) were collected from 27 out of 70 KWD.