Understanding Urea Cycle Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development of a System of High Ornithine and Citrulline Production by a Plant-Derived Lactic Acid Bacterium, Weissella Confusa K-28
Development of a system of high ornithine and citrulline production by a plant-derived lactic acid bacterium, Weissella confusa K-28 㸦᳜≀⏤᮶ங㓟⳦ :HLVVHOODFRQIXVD. ࠾ࡅࡿ ࢜ࣝࢽࢳࣥཬࡧࢩࢺࣝࣜࣥ㧗⏕⏘ࢩࢫࢸ࣒ࡢᵓ⠏㸧 Md Rakhimuzzaman Student ID: D164180 Supervisor: Prof. Masanori Sugiyama Department of Probiotic Science for Preventive Medicine Graduate School of Biomedical and Health Sciences Hiroshima University, Japan 2019 ABBREVIATIONS ADI arginine deiminase AUC area under curve BLAST basic local alignment search tool CK carbamate kinase dNTP deoxyribonucleotide triphosphate DDBJ DNA Data Bank of Japan EDTA ethylenediamine tetra-acetic acid EtOH ethanol EPS exopolysaccharide GABA gamma-aminobutyric acid GRAS generally recognized as safe HPLC high performance liquid chromatography LAB lactic acid bacteria Lb. Lactobacillus OD optical density OTC ornithine carbamoyltransferase PITC phenyl isothiocyanate TAE tris-acetate-ethylenediamine tetra-acetic acid TE tris-ethylenediamine tetra-acetic acid TEA triethylamine Tris tris(hydroxymethyl)aminomethane rDNA ribosomal deoxyribonucleic acid RT-PCR reverse transcription polymerase chain reaction CONTENTS INTRODUCTION 1-17 OBJECTIVES 18 MATERIALS AND METHODS 19-34 RESULTS 35-57 DISCUSSIONS 58-65 CONCLUSION 66-67 ACKNOWLEDGEMENTS 68 REFERENCES 69-84 INTRODUCTION Lactic acid bacteria (LAB) are an order of gram-positive, acid-tolerant, generally nonsporulating, non-respiring, either rod-shaped or spherical microorganisms. Since LAB lack the ability of synthesizing porphyrins (heme, and components of respiratory chains) (Pessione et al, 2010), the bacteria can not generate ATP without external heme supplementation (Pessione et al, 2010). The LAB strains can only obtain ATP by fermentation, usually they generate the ATP from some sugars. Because LAB do not use oxygen for energy production, the bacteria easily grow under anaerobic conditions, but they can also grow in presence of oxygen. -

Metabolic Reprogramming in Prostate Cancer
www.nature.com/bjc REVIEW ARTICLE Metabolic reprogramming in prostate cancer Fahim Ahmad 1,2, Murali Krishna Cherukuri2 and Peter L. Choyke1 Although low risk localised prostate cancer has an excellent prognosis owing to effective treatments, such as surgery, radiation, cryosurgery and hormone therapy, metastatic prostate cancer remains incurable. Existing therapeutic regimens prolong life; however, they are beset by problems of resistance, resulting in poor outcomes. Treatment resistance arises primarily from tumour heterogeneity, altered genetic signatures and metabolic reprogramming, all of which enable the tumour to serially adapt to drugs during the course of treatment. In this review, we focus on alterations in the metabolism of prostate cancer, including genetic signatures and molecular pathways associated with metabolic reprogramming. Advances in our understanding of prostate cancer metabolism might help to explain many of the adaptive responses that are induced by therapy, which might, in turn, lead to the attainment of more durable therapeutic responses. British Journal of Cancer https://doi.org/10.1038/s41416-021-01435-5 BACKGROUND Several tools have been designed to assess metabolism without Cellular metabolism involves complex biochemical processes disturbing the system. Advances in nuclear magnetic resonance through which specific nutrients are consumed. Carbohydrates, and mass spectroscopy have helped to quantify the carbon influx fatty acids and amino acids are the main source of nutrients for of the central metabolic pathways (e.g. TCA cycle, glycolysis and energy homeostasis and macromolecular synthesis. They are the pentose phosphate pathway) in mammalian systems, and this main constituent of core metabolic pathways that can be approach has been instrumental in demonstrating that the classified as anabolic, catabolic or waste producing (Fig. -

Amino Acid Degradation; Urea Cycle
Amino acid degradation; urea cycle Kristina Mlinac Jerković [email protected] In animals, amino acids undergo oxidative degradation in three different metabolic circumstances: 1. During the normal synthesis and degradation of cellular proteins, some amino acids that are released from protein breakdown and are not needed for new protein synthesis undergo oxidative degradation. 2. When a diet is rich in protein and the ingested amino acids exceed the body’s needs for protein synthesis, the surplus is catabolized; amino acids cannot be stored. 3. During starvation or in uncontrolled diabetes mellitus, when carbohydrates are either unavailable or not properly utilized, cellular proteins are used as fuel. Metabolic fates of amino groups - The metabolism of nitrogen-containing molecules such as proteins and nucleic acids differs significantly from that of carbohydrates and lipids. - Whereas the latter molecules can be stored and mobilized as needed for biosynthetic reactions or for energy generation, there is no nitrogen-storing molecule (one exception to this rule is storage protein in seeds). - Organisms must constantly replenish their supply of usable nitrogen to replace organic nitrogen that is lost in catabolism. For example, animals must have a steady supply of amino acids in their diets to replace the nitrogen excreted as urea, uric acid, and other nitrogenous waste products. - Since excess dietary amino acids are neither stored for future use nor excreted, they are converted to common metabolic intermediates such as pyruvate, oxaloacetate, acetyl-CoA, and α-keto-glutarate. - Consequently, amino acids are also precursors of glucose, fatty acids, and ketone bodies and are therefore metabolic fuels. -

Protein Metabolism and It's Disorders
Protein Metabolism and it’s Disorders (Course : M.Sc. Zoology) Course Code: ZOOL 4008 (Biochemistry and Metabolism), Semester –II Dr. Shyam Babu Prasad Assistant Professor Department of Zoology Mahatma Gandhi Central University (MGCU), Motihari-845401 (Bihar) Email: [email protected] Important facts of the Protein Metabolism •Proteins metabolism referred as synthesis of protein from Amino acids (Anabolism) and breakdown of proteins (Catabolism). •In the Unit II, we discussed the synthesis (Anabolism) of various form of the proteins from different amino acids. • Here we mainly focused on the Catabolism of the protein, which is integral part of metabolism of the nitrogen-containing molecules. • Nitrogen enters in to the body in various form of compounds that is present in food, the most important is amino acids (as dietary food). • During Protein/amino acid metabolism (Catabolism), Nitrogen leaves the body as urea, ammonia, and in form of other derivatives. • The Catabolism of the protein depends on the amino acid pool and protein turnover (the rate of protein synthesis and degradation is equal). • The amino acids present in through out of the body (cells, blood, and the extracellular fluids etc) is called amino acid pools. It is maintained by following factors as represented in below diagram: Body protein Dietary degradation Protein 400gm/day 600gm/day Supplied Amino acid pool Depleted Synthesis of Remaining Synthesis of Glucose, Synthesis of amino acids Nitrogen containing Glycogen, the Body burned as compound like Ketone Proteins -
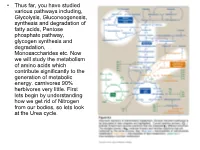
Transaminations and Urea Cycle
• Thus far, you have studied various pathways including, Glycolysis, Gluconeogenesis, synthesis and degradation of fatty acids, Pentose phosphate pathway, glycogen synthesis and degradation, Monosaccharides etc. Now we will study the metabolism of amino acids which contribute significantly to the generation of metabolic energy. carnivores 90% herbivores very little. First lets begin by understanding how we get rid of Nitrogen from our bodies, so lets look at the Urea cycle. 1 α Amino group keeps amino acids safe from oxidative breakdown. Removal of this group is essential for energy production it is an obligatory step for catabolism. Nitrogen can then be incorporated or excreted.This is done through transamination or oxidative deamination, provide amonia and aspartate sources for the Urea cycle. Transamination is funneled through glutamate. This is done through the transfer of the α amino group to α ketoglutarate. Product α-keto acid and glutamate. Alanine to pyruvate and aspartate to oxaloacetate all amino acids except lysine and threonina (lose the α amino through deamination. Glutamate dehydrogenase the oxidative deamination Glutamate only amino acid that undergoes rapid oxidative deamination (glutamate dehydrogenase NH3 + NADH), where amino groups are released as ammonia. After a meal rich in protein the levels of glutamate in liver are increased favoring amino acid degradation and formation of ammonia. Activators of this enzyme are ADP and GDP while ATP and GTP are inhibitors. This reaction occurs in the mitochondria. Ammonia can be transported to the liver from other tissues through two mechanisms that reduce the toxicity of ammonia. One through Glutamine (most tissues through, glutamine synthetase). In the liver the glutamine is converted back to glutamate (Fig. -

ATP Structure and Function
ATP: Universal Currency of Cellular Energy All living things including plants, animals, birds, insects, humans need energy for the proper functioning of cells, tissues and other organ systems. As we are aware that green plants, obtain their energy from the sunlight, and animals get their energy by feeding on these plants. Energy acts as a source of fuel. We, humans, gain energy from the food we eat, but how are the energy produced and stored in our body. A living cell cannot store significant amounts of free energy. Excess free energy would result in an increase of heat in the cell, which would result in excessive thermal motion that could damage and then destroy the cell. Rather, a cell must be able to handle that energy in a way that enables the cell to store energy safely and release it for use only as needed. Living cells accomplish this by using the compound adenosine triphosphate (ATP). ATP is often called the “energy currency” of the cell, and, like currency, this versatile compound can be used to fill any energy need of the cell. How? It functions similarly to a rechargeable battery. When ATP is broken down, usually by the removal of its terminal phosphate group, energy is released. The energy is used to do work by the cell, usually by the released phosphate binding to another molecule, activating it. For example, in the mechanical work of muscle contraction, ATP supplies the energy to move the contractile muscle proteins. Recall the active transport work of the sodium-potassium pump in cell membranes. -

Laboratory Diagnostic Approaches in Metabolic Disorders
470 Review Article on Inborn Errors of Metabolism Page 1 of 14 Laboratory diagnostic approaches in metabolic disorders Ruben Bonilla Guerrero1, Denise Salazar2, Pranoot Tanpaiboon2,3 1Formerly Quest Diagnostics, Inc., Ruben Bonilla Guerrero, Rancho Santa Margarita, CA, USA; 2Quest Diagnostics, Inc., Denise Salazar and Pranoot Tanpaiboon, San Juan Capistrano, CA, USA; 3Genetics and Metabolism, Children’s National Rare Disease Institute, Washington, DC, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: R Bonilla Guerrero; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: None; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Ruben Bonilla Guerrero. Formerly Quest Diagnostics, Inc., Ruben Bonilla Guerrero, 508 Sable, Rancho Santa Margarita, CA 92688, USA. Email: [email protected]. Abstract: The diagnosis of inborn errors of metabolism (IEM) takes many forms. Due to the implementation and advances in newborn screening (NBS), the diagnosis of many IEM has become relatively easy utilizing laboratory biomarkers. For the majority of IEM, early diagnosis prevents the onset of severe clinical symptoms, thus reducing morbidity and mortality. However, due to molecular, biochemical, and clinical variability of IEM, not all disorders included in NBS programs will be detected and diagnosed by screening alone. This article provides a general overview and simplified guidelines for the diagnosis of IEM in patients with and without an acute metabolic decompensation, with early or late onset of clinical symptoms. The proper use of routine laboratory results in the initial patient assessment is also discussed, which can help guide efficient ordering of specialized laboratory tests to confirm a potential diagnosis and initiate treatment as soon as possible. -

Amino Acid Catabolism: Urea Cycle the Urea Bi-Cycle Two Issues
BI/CH 422/622 OUTLINE: OUTLINE: Protein Degradation (Catabolism) Digestion Amino-Acid Degradation Inside of cells Urea Cycle – dealing with the nitrogen Protein turnover Ubiquitin Feeding the Urea Cycle Activation-E1 Glucose-Alanine Cycle Conjugation-E2 Free Ammonia Ligation-E3 Proteosome Glutamine Amino-Acid Degradation Glutamate dehydrogenase Ammonia Overall energetics free Dealing with the carbon transamination-mechanism to know Seven Families Urea Cycle – dealing with the nitrogen 1. ADENQ 5 Steps 2. RPH Carbamoyl-phosphate synthetase oxidase Ornithine transcarbamylase one-carbon metabolism Arginino-succinate synthetase THF Arginino-succinase SAM Arginase 3. GSC Energetics PLP uses Urea Bi-cycle 4. MT – one carbon metabolism 5. FY – oxidases Amino Acid Catabolism: Urea Cycle The Urea Bi-Cycle Two issues: 1) What to do with the fumarate? 2) What are the sources of the free ammonia? a-ketoglutarate a-amino acid Aspartate transaminase transaminase a-keto acid Glutamate 1 Amino Acid Catabolism: Urea Cycle The Glucose-Alanine Cycle • Vigorously working muscles operate nearly anaerobically and rely on glycolysis for energy. a-Keto acids • Glycolysis yields pyruvate. – If not eliminated (converted to acetyl- CoA), lactic acid will build up. • If amino acids have become a fuel source, this lactate is converted back to pyruvate, then converted to alanine for transport into the liver. Excess Glutamate is Metabolized in the Mitochondria of Hepatocytes Amino Acid Catabolism: Urea Cycle Excess glutamine is processed in the intestines, kidneys, and liver. (deaminating) (N,Q,H,S,T,G,M,W) OAA à Asp Glutamine Synthetase This costs another ATP, bringing it closer to 5 (N,Q,H,S,T,G,M,W) 29 N 2 Amino Acid Catabolism: Urea Cycle Excess glutamine is processed in the intestines, kidneys, and liver. -

Unit 8 Amino Acid and Nucleotide Metabolism
UNIT 8 AMINO ACID AND NUCLEOTIDE METABOLISM 8.1 Introduction 8.2 Amino Acid Metabolism 8 2 1 Transamination Reaction 8 2.2 Deamination Reaction 8.2.3 Urea Cycle 8 2.4 Metabolism of Carbon Skeletons of Amino Acids 8 2.5 Biosynthesis of Nonessential Amino Acids 8 2.6 Synthesis of Specialized Products from Amino Acids 8 2.7 Decarboxylation Reaction and Biogenic Amines 8.2.8 Non-protein Functions of Amino Acids 8.3 Nucleotide Metabolism 8.3.1 Purine Nucleotide Synthesis- De Novo Synthesis 8.3.2 Salvage Pathway for Purines 8.3.3 Degradation of Purine Nucleotides 8.3.4 Pyrimidine Synthesis 8 3.5 Regulation of Deoxyribonucleotide Synthesis 8.4 Let Us Sum Up 8.5 Glossary I. 8.6 Answers to Check Your Progress Exercises 8.1 INTRODUCTION In Unit 2, we studied about the chemistry of proteins and amino acids. We studied that the amino acids are used for protein synthesis. Subsequently, in Unit 5 we studied about the digestion, absorption and transport of proteins and amino acids. We learnt that proteins do not immediately diffuse into the surrounding tissues of the blood stream, but first undergo a series of steps of biochemical reactions in the digestive tract. These reactions reduce the protein into its individual amino acids which are rapidly absorbed into the blood stream. Amino acids contain nitrogen, which cannot be stored, and therefore amino acids', which are in excess of biosynthetic needs of the cell, are degraded. This involves the removal of a-amino groups (amino groups attached to carbon atom next to the carboxyl carbon) by two processes. -

Hormonal Regulation of Two Urea-Cycle Enzymes in Cultured
Biochem. J. (1983) 216, 281-285 281 Printed in Great Britain Hormonal regulation oftwo urea-cycle enzymes in cultured foetal hepatocytes Annie HUSSON, Mimouna BOUAZZA, Catherine BUQUET and Rene VAILLANT Laboratoire d'Endocrinologie ERA 891, Faculte des Sciences et des Techniques, 76130 Mont-Saint-A ignan, France (Received 5 April 1983/Accepted 11 July 1983) Foetal-rat hepatocytes were cultured in primary monolayer culture, and activity changes of argininosuccinate synthetase (ASS, EC 6.3.4.5) and argininosuccinase (ASL, EC 4.3.2.1) were followed under defined hormone conditions. In hormone-free medium, cultured cells maintained the enzyme activities at values equal to those of freshly isolated cells for at least 3 days. Continuous addition of dexamethasone produced the development of the two enzyme activities, but only after the first 20h of culture. Under these conditions, urea production by the foetal hepatocytes was concomitantly increased in the culture medium. Pretreatment with dexamethasone for 20h was sufficient to produce the development of ASL activity within the 2 following days. Introduced alone, glucagon induced an increase of ASL activity, but did not affect the ASS activity. The most powerful stimulation of ASS and ASL could be observed in cultured hepatocytes if glucagon and dexamethasone were added simultaneously or sequentially. These results indicated that the development of the receptor complex for the induction of urea-cycle enzymes appears early before birth and established that glucocorticoids amplify the glucagon stimulation of these enzyme activities during foetal life. During development of the foetal rat liver, the To obtain more definite information, we have used appearance of a number of enzymes has been shown primary culture of foetal liver cells, which has been to result from hormone action (Jost & Picon, 1970; shown to be a very attractive model for studying Greengard, 1970). -

Urea Cycle Disorders: a Life-Threatening Yet Treatable Cause of Metabolic Encephalopathy in Adults
NEUROLOGICAL RARITIES Pract Neurol: first published as 10.1136/practneurol-2014-000916 on 14 August 2014. Downloaded from Urea cycle disorders: a life-threatening yet treatable cause of metabolic encephalopathy in adults Nicholas F Blair,1 Philip D Cremer,1 Michel C Tchan2 1Department of Neurology, Royal ABSTRACT postoperative pain. She had no previous North Shore Hospital, Sydney, Urea cycle disorders are inborn errors of mood disorder or other psychiatric condi- Australia 2Adult Genetic Metabolic metabolism that, in rare cases, can present for tion. There was no recent alcohol intake Disorders Service, Westmead the first time in adulthood. We report a or history of substance abuse. Hospital, Sydney, Australia perplexing presentation in a woman 4 days Her initial blood results showed mul- postpartum of bizarre and out-of-character Correspondence to tiple mild abnormalities (table 1), many Dr Michel C Tchan, Department behaviour interspersed with periods of complete of which could be explained by recent of Genetic Medicine, Westmead normality. Without any focal neurological signs pregnancy; in particular, there was no Hospital, Hawkesbury Road, or abnormality on initial investigations, the renal or hepatic dysfunction, other than Westmead NSW 2145, Australia; [email protected] diagnosis became clear with the finding of a mildly elevated serum alkaline phosphat- significantly elevated plasma ammonia level, just ase, which may have been due to placen- Accepted 24 June 2014 as she began to deteriorate rapidly. She tal production. Her CT scan of head was Published Online First improved following intravenous dextrose and normal. CSF protein was mildly elevated 14 August 2014 lipid emulsion, together with sodium benzoate, at 0.58 g/L (0.15–0.45) with normal CSF arginine and a protein-restricted diet. -

Metabolism Citric Acid Cycle
INTRODUCTION TO CELL METABOLISM CITRIC ACID CYCLE Prof. MUDr. Jiří Kraml, DrSc. Institute of Medical Biochemistry, Charles Univ., First Fac.Med. METABOLISM = turnover of substances in the organism - chemical (compounds) feature - reactions - anabolic - catabolic - amphibolic - energetic feature - reactions exergonic: ∆G<0 ∆U(∆G) ——> work + heat - endergonic: ∆G>0 Anabolic (association, biosynthetic reactions) are endergonic, catabolic (dissociation) reactions are exergonic, amphibolic reactions serve both biosyntheses and biodegradations (citrate Krebs cycle). Endergonic reactions include : -biosyntheses (reduction, condensation), -osmotic work (concentration-gradients, ion-gradients), -mechanical work (muscle contraction - conformation changes of contractile proteins). Exergonic reactions include : - oxidation (loss of electrons, loss of H atoms, entry of O atoms into the molecule), - hydrolytic reactions, esp. of anhydride bonds (e.g. diphosphates, triphosphates). ∆G of oxidation-reduction reaction (exergonic): n = moles of electrons; pH= 7; -1 -1 ∆G´ = - n . F . ∆E´h ; F = 96.5 kJ . V . mol ; ∆E´h = difference in redox-potentials in V. ∆G of osmotic work (endergonic): ∆G = n . R . T . ln c2 / c1 ; c2>c1 ; n = moles of trans- o -1 ferred particles; for 37 C : ∆G = n . 5.9 . log c2 / c1 [kJ . mol ] Elektrochemical potential (gradient) for ions ( n=1): ∆G = R . T . ln c2 / c1 + Z . F . ∆V ; Z = charge of particles; ∆V = membrane potential in V 1 OXIDATION-REDUCTION POTENTIAL (REDOX POTENTIAL) E´h is a quantity measuring and expressing the power of a system to donate or accept electrons. A system donating electrons functions as a reducing half-cell and has a more negative redox potential ; a system accepting electrons operates as an oxidizing half-cell and possesses a more positive redox potential.