Anaesthesia Recommendations for Urea Cycle Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hyperammonemia in Review: Pathophysiology, Diagnosis, and Treatment
Pediatr Nephrol DOI 10.1007/s00467-011-1838-5 EDUCATIONAL REVIEW Hyperammonemia in review: pathophysiology, diagnosis, and treatment Ari Auron & Patrick D. Brophy Received: 23 September 2010 /Revised: 9 January 2011 /Accepted: 12 January 2011 # IPNA 2011 Abstract Ammonia is an important source of nitrogen and is the breakdown and catabolism of dietary and bodily proteins, required for amino acid synthesis. It is also necessary for respectively. In healthy individuals, amino acids that are not normal acid-base balance. When present in high concentra- needed for protein synthesis are metabolized in various tions, ammonia is toxic. Endogenous ammonia intoxication chemical pathways, with the rest of the nitrogen waste being can occur when there is impaired capacity of the body to converted to urea. Ammonia is important for normal animal excrete nitrogenous waste, as seen with congenital enzymatic acid-base balance. During exercise, ammonia is produced in deficiencies. A variety of environmental causes and medica- skeletal muscle from deamination of adenosine monophos- tions may also lead to ammonia toxicity. Hyperammonemia phate and amino acid catabolism. In the brain, the latter refers to a clinical condition associated with elevated processes plus the activity of glutamate dehydrogenase ammonia levels manifested by a variety of symptoms and mediate ammonia production. After formation of ammonium signs, including significant central nervous system (CNS) from glutamine, α-ketoglutarate, a byproduct, may be abnormalities. Appropriate and timely management requires a degraded to produce two molecules of bicarbonate, which solid understanding of the fundamental pathophysiology, are then available to buffer acids produced by dietary sources. differential diagnosis, and treatment approaches available. -

Development of a System of High Ornithine and Citrulline Production by a Plant-Derived Lactic Acid Bacterium, Weissella Confusa K-28
Development of a system of high ornithine and citrulline production by a plant-derived lactic acid bacterium, Weissella confusa K-28 㸦᳜≀⏤᮶ங㓟⳦ :HLVVHOODFRQIXVD. ࠾ࡅࡿ ࢜ࣝࢽࢳࣥཬࡧࢩࢺࣝࣜࣥ㧗⏕⏘ࢩࢫࢸ࣒ࡢᵓ⠏㸧 Md Rakhimuzzaman Student ID: D164180 Supervisor: Prof. Masanori Sugiyama Department of Probiotic Science for Preventive Medicine Graduate School of Biomedical and Health Sciences Hiroshima University, Japan 2019 ABBREVIATIONS ADI arginine deiminase AUC area under curve BLAST basic local alignment search tool CK carbamate kinase dNTP deoxyribonucleotide triphosphate DDBJ DNA Data Bank of Japan EDTA ethylenediamine tetra-acetic acid EtOH ethanol EPS exopolysaccharide GABA gamma-aminobutyric acid GRAS generally recognized as safe HPLC high performance liquid chromatography LAB lactic acid bacteria Lb. Lactobacillus OD optical density OTC ornithine carbamoyltransferase PITC phenyl isothiocyanate TAE tris-acetate-ethylenediamine tetra-acetic acid TE tris-ethylenediamine tetra-acetic acid TEA triethylamine Tris tris(hydroxymethyl)aminomethane rDNA ribosomal deoxyribonucleic acid RT-PCR reverse transcription polymerase chain reaction CONTENTS INTRODUCTION 1-17 OBJECTIVES 18 MATERIALS AND METHODS 19-34 RESULTS 35-57 DISCUSSIONS 58-65 CONCLUSION 66-67 ACKNOWLEDGEMENTS 68 REFERENCES 69-84 INTRODUCTION Lactic acid bacteria (LAB) are an order of gram-positive, acid-tolerant, generally nonsporulating, non-respiring, either rod-shaped or spherical microorganisms. Since LAB lack the ability of synthesizing porphyrins (heme, and components of respiratory chains) (Pessione et al, 2010), the bacteria can not generate ATP without external heme supplementation (Pessione et al, 2010). The LAB strains can only obtain ATP by fermentation, usually they generate the ATP from some sugars. Because LAB do not use oxygen for energy production, the bacteria easily grow under anaerobic conditions, but they can also grow in presence of oxygen. -

Metabolic Reprogramming in Prostate Cancer
www.nature.com/bjc REVIEW ARTICLE Metabolic reprogramming in prostate cancer Fahim Ahmad 1,2, Murali Krishna Cherukuri2 and Peter L. Choyke1 Although low risk localised prostate cancer has an excellent prognosis owing to effective treatments, such as surgery, radiation, cryosurgery and hormone therapy, metastatic prostate cancer remains incurable. Existing therapeutic regimens prolong life; however, they are beset by problems of resistance, resulting in poor outcomes. Treatment resistance arises primarily from tumour heterogeneity, altered genetic signatures and metabolic reprogramming, all of which enable the tumour to serially adapt to drugs during the course of treatment. In this review, we focus on alterations in the metabolism of prostate cancer, including genetic signatures and molecular pathways associated with metabolic reprogramming. Advances in our understanding of prostate cancer metabolism might help to explain many of the adaptive responses that are induced by therapy, which might, in turn, lead to the attainment of more durable therapeutic responses. British Journal of Cancer https://doi.org/10.1038/s41416-021-01435-5 BACKGROUND Several tools have been designed to assess metabolism without Cellular metabolism involves complex biochemical processes disturbing the system. Advances in nuclear magnetic resonance through which specific nutrients are consumed. Carbohydrates, and mass spectroscopy have helped to quantify the carbon influx fatty acids and amino acids are the main source of nutrients for of the central metabolic pathways (e.g. TCA cycle, glycolysis and energy homeostasis and macromolecular synthesis. They are the pentose phosphate pathway) in mammalian systems, and this main constituent of core metabolic pathways that can be approach has been instrumental in demonstrating that the classified as anabolic, catabolic or waste producing (Fig. -

Inherited Metabolic Disease
Inherited metabolic disease Dr Neil W Hopper SRH Areas for discussion • Introduction to IEMs • Presentation • Initial treatment and investigation of IEMs • Hypoglycaemia • Hyperammonaemia • Other presentations • Management of intercurrent illness • Chronic management Inherited Metabolic Diseases • Result from a block to an essential pathway in the body's metabolism. • Huge number of conditions • All rare – very rare (except for one – 1:500) • Presentation can be non-specific so index of suspicion important • Mostly AR inheritance – ask about consanguinity Incidence (W. Midlands) • Amino acid disorders (excluding phenylketonuria) — 18.7 per 100,000 • Phenylketonuria — 8.1 per 100,000 • Organic acidemias — 12.6 per 100,000 • Urea cycle diseases — 4.5 per 100,000 • Glycogen storage diseases — 6.8 per 100,000 • Lysosomal storage diseases — 19.3 per 100,000 • Peroxisomal disorders — 7.4 per 100,000 • Mitochondrial diseases — 20.3 per 100,000 Pathophysiological classification • Disorders that result in toxic accumulation – Disorders of protein metabolism (eg, amino acidopathies, organic acidopathies, urea cycle defects) – Disorders of carbohydrate intolerance – Lysosomal storage disorders • Disorders of energy production, utilization – Fatty acid oxidation defects – Disorders of carbohydrate utilization, production (ie, glycogen storage disorders, disorders of gluconeogenesis and glycogenolysis) – Mitochondrial disorders – Peroxisomal disorders IMD presentations • ? IMD presentations • Screening – MCAD, PKU • Progressive unexplained neonatal -

Summary Current Practices Report
18/10/2011 EU Tender “Evaluation of population newborn screening practices for rare disorders in Member States of the European Union” Short Executive Summary of the Report on the practices of newborn screening for rare disorders implemented in Member States of the European Union, Candidate, Potential Candidate and EFTA Countries Authors: Peter Burgard1, Martina Cornel2, Francesco Di Filippo4, Gisela Haege1, Georg F. Hoffmann1, Martin Lindner1, J. Gerard Loeber3, Tessel Rigter2, Kathrin Rupp1, 4 Domenica Taruscio4,Luciano Vittozzi , Stephanie Weinreich2 1 Department of Pediatrics , University Hospital - Heidelberg (DE) 2 VU University Medical Centre - Amsterdam (NL) 3 RIVM - Bilthoven (NL) 4 National Centre for Rare Diseases - Rome (IT) The opinions expressed in this document are those of the Contractor only and do not represent the official position of the Executive Agency for Health and Consumers. This work is funded by the European Union with a grant of Euro 399755 (Contract number 2009 62 06 of the Executive Agency for Health and Consumers) 1 18/10/2011 Abbreviations 3hmg 3-Hydroxy-3-methylglutaric aciduria 3mcc 3-Methylcrotonyl-CoA carboxylase deficiency/3-Methylglutacon aciduria/2-methyl-3-OH- butyric aciduria AAD Disorders of amino acid metabolism arg Argininemia asa Argininosuccinic aciduria bio Biotinidase deficiency bkt Beta-ketothiolase deficiency btha S, beta 0-thalassemia cah Congenital adrenal hyperplasia cf Cystic fibrosis ch Primary congenital hypothyroidism citI Citrullinemia type I citII Citrullinemia type II cpt I Carnitin -

Argininosuccinate Lyase Deficiency
©American College of Medical Genetics and Genomics GENETEST REVIEW Argininosuccinate lyase deficiency Sandesh C.S. Nagamani, MD1, Ayelet Erez, MD, PhD1 and Brendan Lee, MD, PhD1,2 The urea cycle consists of six consecutive enzymatic reactions that citrulline together with elevated argininosuccinic acid in the plasma convert waste nitrogen into urea. Deficiencies of any of these enzymes or urine. Molecular genetic testing of ASL and assay of ASL enzyme of the cycle result in urea cycle disorders (UCDs), a group of inborn activity are helpful when the biochemical findings are equivocal. errors of hepatic metabolism that often result in life-threatening However, there is no correlation between the genotype or enzyme hyperammonemia. Argininosuccinate lyase (ASL) catalyzes the activity and clinical outcome. Treatment of acute metabolic decom- fourth reaction in this cycle, resulting in the breakdown of arginino- pensations with hyperammonemia involves discontinuing oral pro- succinic acid to arginine and fumarate. ASL deficiency (ASLD) is the tein intake, supplementing oral intake with intravenous lipids and/ second most common UCD, with a prevalence of ~1 in 70,000 live or glucose, and use of intravenous arginine and nitrogen-scavenging births. ASLD can manifest as either a severe neonatal-onset form therapy. Dietary restriction of protein and dietary supplementation with hyperammonemia within the first few days after birth or as a with arginine are the mainstays in long-term management. Ortho- late-onset form with episodic hyperammonemia and/or long-term topic liver transplantation (OLT) is best considered only in patients complications that include liver dysfunction, neurocognitive deficits, with recurrent hyperammonemia or metabolic decompensations and hypertension. -

What Disorders Are Screened for by the Newborn Screen?
What disorders are screened for by the newborn screen? Endocrine Disorders The endocrine system is important to regulate the hormones in our bodies. Hormones are special signals sent to various parts of the body. They control many things such as growth and development. The goal of newborn screening is to identify these babies early so that treatment can be started to keep them healthy. To learn more about these specific disorders please click on the name of the disorder below: English: Congenital Adrenal Hyperplasia Esapnol Hiperplasia Suprarrenal Congenital - - http://www.newbornscreening.info/Parents/otherdisorders/CAH.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/CAH.html - Congenital Hypothyroidism (Hipotiroidismo Congénito) - http://www.newbornscreening.info/Parents/otherdisorders/CH.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/CH.html Hematologic Conditions Hemoglobin is a special part of our red blood cells. It is important for carrying oxygen to the parts of the body where it is needed. When people have problems with their hemoglobin they can have intense pain, and they often get sick more than other children. Over time, the lack of oxygen to the body can cause damage to the organs. The goal of newborn screening is to identify babies with these conditions so that they can get early treatment to help keep them healthy. To learn more about these specific disorders click here (XXX). - Sickle Cell Anemia (Anemia de Célula Falciforme) - http://www.newbornscreening.info/Parents/otherdisorders/SCD.html - http://www.newbornscreening.info/spanish/parent/Other_disorder/SCD.html - SC Disease (See Previous Link) - Sickle Beta Thalassemia (See Previous Link) Enzyme Deficiencies Enzymes are special proteins in our body that allow for chemical reactions to take place. -

Amino Acid Degradation; Urea Cycle
Amino acid degradation; urea cycle Kristina Mlinac Jerković [email protected] In animals, amino acids undergo oxidative degradation in three different metabolic circumstances: 1. During the normal synthesis and degradation of cellular proteins, some amino acids that are released from protein breakdown and are not needed for new protein synthesis undergo oxidative degradation. 2. When a diet is rich in protein and the ingested amino acids exceed the body’s needs for protein synthesis, the surplus is catabolized; amino acids cannot be stored. 3. During starvation or in uncontrolled diabetes mellitus, when carbohydrates are either unavailable or not properly utilized, cellular proteins are used as fuel. Metabolic fates of amino groups - The metabolism of nitrogen-containing molecules such as proteins and nucleic acids differs significantly from that of carbohydrates and lipids. - Whereas the latter molecules can be stored and mobilized as needed for biosynthetic reactions or for energy generation, there is no nitrogen-storing molecule (one exception to this rule is storage protein in seeds). - Organisms must constantly replenish their supply of usable nitrogen to replace organic nitrogen that is lost in catabolism. For example, animals must have a steady supply of amino acids in their diets to replace the nitrogen excreted as urea, uric acid, and other nitrogenous waste products. - Since excess dietary amino acids are neither stored for future use nor excreted, they are converted to common metabolic intermediates such as pyruvate, oxaloacetate, acetyl-CoA, and α-keto-glutarate. - Consequently, amino acids are also precursors of glucose, fatty acids, and ketone bodies and are therefore metabolic fuels. -

Protein Metabolism and It's Disorders
Protein Metabolism and it’s Disorders (Course : M.Sc. Zoology) Course Code: ZOOL 4008 (Biochemistry and Metabolism), Semester –II Dr. Shyam Babu Prasad Assistant Professor Department of Zoology Mahatma Gandhi Central University (MGCU), Motihari-845401 (Bihar) Email: [email protected] Important facts of the Protein Metabolism •Proteins metabolism referred as synthesis of protein from Amino acids (Anabolism) and breakdown of proteins (Catabolism). •In the Unit II, we discussed the synthesis (Anabolism) of various form of the proteins from different amino acids. • Here we mainly focused on the Catabolism of the protein, which is integral part of metabolism of the nitrogen-containing molecules. • Nitrogen enters in to the body in various form of compounds that is present in food, the most important is amino acids (as dietary food). • During Protein/amino acid metabolism (Catabolism), Nitrogen leaves the body as urea, ammonia, and in form of other derivatives. • The Catabolism of the protein depends on the amino acid pool and protein turnover (the rate of protein synthesis and degradation is equal). • The amino acids present in through out of the body (cells, blood, and the extracellular fluids etc) is called amino acid pools. It is maintained by following factors as represented in below diagram: Body protein Dietary degradation Protein 400gm/day 600gm/day Supplied Amino acid pool Depleted Synthesis of Remaining Synthesis of Glucose, Synthesis of amino acids Nitrogen containing Glycogen, the Body burned as compound like Ketone Proteins -
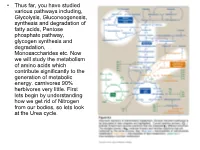
Transaminations and Urea Cycle
• Thus far, you have studied various pathways including, Glycolysis, Gluconeogenesis, synthesis and degradation of fatty acids, Pentose phosphate pathway, glycogen synthesis and degradation, Monosaccharides etc. Now we will study the metabolism of amino acids which contribute significantly to the generation of metabolic energy. carnivores 90% herbivores very little. First lets begin by understanding how we get rid of Nitrogen from our bodies, so lets look at the Urea cycle. 1 α Amino group keeps amino acids safe from oxidative breakdown. Removal of this group is essential for energy production it is an obligatory step for catabolism. Nitrogen can then be incorporated or excreted.This is done through transamination or oxidative deamination, provide amonia and aspartate sources for the Urea cycle. Transamination is funneled through glutamate. This is done through the transfer of the α amino group to α ketoglutarate. Product α-keto acid and glutamate. Alanine to pyruvate and aspartate to oxaloacetate all amino acids except lysine and threonina (lose the α amino through deamination. Glutamate dehydrogenase the oxidative deamination Glutamate only amino acid that undergoes rapid oxidative deamination (glutamate dehydrogenase NH3 + NADH), where amino groups are released as ammonia. After a meal rich in protein the levels of glutamate in liver are increased favoring amino acid degradation and formation of ammonia. Activators of this enzyme are ADP and GDP while ATP and GTP are inhibitors. This reaction occurs in the mitochondria. Ammonia can be transported to the liver from other tissues through two mechanisms that reduce the toxicity of ammonia. One through Glutamine (most tissues through, glutamine synthetase). In the liver the glutamine is converted back to glutamate (Fig. -

ATP Structure and Function
ATP: Universal Currency of Cellular Energy All living things including plants, animals, birds, insects, humans need energy for the proper functioning of cells, tissues and other organ systems. As we are aware that green plants, obtain their energy from the sunlight, and animals get their energy by feeding on these plants. Energy acts as a source of fuel. We, humans, gain energy from the food we eat, but how are the energy produced and stored in our body. A living cell cannot store significant amounts of free energy. Excess free energy would result in an increase of heat in the cell, which would result in excessive thermal motion that could damage and then destroy the cell. Rather, a cell must be able to handle that energy in a way that enables the cell to store energy safely and release it for use only as needed. Living cells accomplish this by using the compound adenosine triphosphate (ATP). ATP is often called the “energy currency” of the cell, and, like currency, this versatile compound can be used to fill any energy need of the cell. How? It functions similarly to a rechargeable battery. When ATP is broken down, usually by the removal of its terminal phosphate group, energy is released. The energy is used to do work by the cell, usually by the released phosphate binding to another molecule, activating it. For example, in the mechanical work of muscle contraction, ATP supplies the energy to move the contractile muscle proteins. Recall the active transport work of the sodium-potassium pump in cell membranes. -

Laboratory Diagnostic Approaches in Metabolic Disorders
470 Review Article on Inborn Errors of Metabolism Page 1 of 14 Laboratory diagnostic approaches in metabolic disorders Ruben Bonilla Guerrero1, Denise Salazar2, Pranoot Tanpaiboon2,3 1Formerly Quest Diagnostics, Inc., Ruben Bonilla Guerrero, Rancho Santa Margarita, CA, USA; 2Quest Diagnostics, Inc., Denise Salazar and Pranoot Tanpaiboon, San Juan Capistrano, CA, USA; 3Genetics and Metabolism, Children’s National Rare Disease Institute, Washington, DC, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: R Bonilla Guerrero; (III) Provision of study materials or patients: All authors; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: None; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors. Correspondence to: Ruben Bonilla Guerrero. Formerly Quest Diagnostics, Inc., Ruben Bonilla Guerrero, 508 Sable, Rancho Santa Margarita, CA 92688, USA. Email: [email protected]. Abstract: The diagnosis of inborn errors of metabolism (IEM) takes many forms. Due to the implementation and advances in newborn screening (NBS), the diagnosis of many IEM has become relatively easy utilizing laboratory biomarkers. For the majority of IEM, early diagnosis prevents the onset of severe clinical symptoms, thus reducing morbidity and mortality. However, due to molecular, biochemical, and clinical variability of IEM, not all disorders included in NBS programs will be detected and diagnosed by screening alone. This article provides a general overview and simplified guidelines for the diagnosis of IEM in patients with and without an acute metabolic decompensation, with early or late onset of clinical symptoms. The proper use of routine laboratory results in the initial patient assessment is also discussed, which can help guide efficient ordering of specialized laboratory tests to confirm a potential diagnosis and initiate treatment as soon as possible.