PD-1: Its Discovery, Involvement in Cancer Immunotherapy, and Beyond
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Annual Report Fy 2018 Human Frontier Science Program Organization
APRIL 2017 APRIL 2018 — MARCH 2019 ANNUAL REPORT FY 2018 HUMAN FRONTIER SCIENCE PROGRAM ORGANIZATION The Human Frontier Science Program Organization (HFSPO) is unique, supporting international collaboration to undertake innovative, risky, basic research at the frontier of the life sciences. Special emphasis is given to the support and training of independent young investigators, beginning at the postdoctoral level. The Program is implemented by an international organisation, supported financially by Australia, Canada, France, Germany, India, Italy, Japan, the Republic of Korea, New Zealand, Norway, Singapore, Switzerland, the United Kingdom of Great Britain and Nothern Ireland, the United States of America, and the European Commission. Since 1990, over 7000 researchers from more than 70 countries have been supported. Of these, 28 HFSP awardees have gone on to receive the Nobel Prize. 2 The following documents are available on the HFSP website www.hfsp.org: Joint Communiqués (Tokyo 1992, Washington 1997, Berlin 2002, Bern 2004, Ottawa 2007, Canberra 2010, Brussels 2013, London 2016): https://www.hfsp.org/about/governance/membership Statutes of the International Human Frontier Science Program Organization: https://www.hfsp.org/about/governance/hfspo-statutes Guidelines for the participation of new members in HFSPO: https://www.hfsp.org/about/governance/membership General reviews of the HFSP (1996, 2001, 2006-2007, 2010, 2018): https://www.hfsp.org/about/strategy/reviews Updated and previous lists of awards, including titles and abstracts: -

October 11, 1994, NIH Record, Vol. XLVI, No. 21
October l 1, 1994 Vol. XLVI No. 21 "Still U.S. Department of Health The Second and Human Services Best Thing About Payday" National Institutes of Health Dunbar To Give First Pittman Lecture, Oct. 26 By Sara Byars scientist recognized internarionally for A her pioneering work in contraceptive vaccines has been selected to deliver the first Margaret Pittman Lecture, a new NIH series chat honors outstanding women scientists. Dr. Bonnie S. Dunbar, professor of cell . :/ biology and obstetrics and gynecology at Baylor College of Medicine, will speak on "New ':'' .:' f ,; ... .. Fromiers in Reproductive Biology and I !- J ~ , • I , , ' f ., ' / , , Contraceptive Vaccines" at 3 p.m. on 0cc. 26 ' • /} : 4 t ., : in Masur Auditorium, Bldg. I 0 . ' ' . ! • ' I I I • Guiding che development of che Pittman • ' ' ,• ,: k'I I ' I lectureship series is the NIH women scientists J: • ! • •'I I It♦ l'- : ,4 • • •· ... / ;~ ~ advisory committee, a group char advises At the NIH Research Festival 1994 poster session, Dr. Lynn Hudson (r), chief. molecular genetics section scientific directors on matters pertaining co rhe ofNJNDS' Laboratory ofViral and Molecular Pathogenesis, stops by to view the work ofDr. Rosemary role of women scientists at NIH. Wong, one ofthe authors who works in NIDDK's Molemlar, Cellular and Endocrinology Branch. See This lectureship honors Or. Margaret additional coverage ofthe event on Pages 6-7. Pittman, rhe first woman co hold the position (See PITTMAN LECTURE, Page 2) NIGMS Reorganizes, Moves to Natcher Bldg. is month, che N acional Institute of General Medical Sciences is undergoing a reorganiza Vitetta Is NIAID's 1994 tion and a move to che new William H. -

Science, Technology and Education News from Taiwan
TRADE OFFICE OF SWISS INDUSTRIES Press Review Taiwan Science, Technology and Education Trade Office of Swiss Industries, Taipei, October 131, 2017 Science/Technology ............................................................................................................................................ 2 Taiwan wins 50 medals, 6 special prizes at Ukraine invention show (Focus Taiwan, 1.10.2017) ................... 2 The story behind TSMC's Morris Chang (Focus Taiwan, 2.10.2017)............................................................... 2 Taiwan, India foster closer ties on technological development (Taiwan Today, 13.10.2017) ........................... 2 Tang Prize winner develops new genome editing techniques (Focus Taiwan, 13.10.2017) ............................ 2 Taiwan contributes to historic astronomy research (Focus Taiwan, 17.10.2017) ............................................ 2 Plans for second set of FormoSat-7 satellites scrapped: NSPO (Focus Taiwan, 20.10.2017) ........................ 2 Taiwan aiming to boost cooperation with U.S. through training program (Focus Taiwan, 20.10.2017) ........... 2 3 Taiwan tech firms feature in Forbes top regarded company list (Focus Taiwan, 21.10.2017) ...................... 2 Science ministry to invest NT$4 billion on AI training (Focus Taiwan, 24.10.2017) ......................................... 2 Qualcomm suspends 5G cooperation with Taiwan's ITRI (Focus Taiwan, 25.10.2017) .................................. 3 Four Taiwan cities, counties listed in 2018 Smart21 Communities (Focus -

Mage, Rose G. 2018 Dr
Mage, Rose G. 2018 Dr. Rose G. Mage Oral History Download the PDF: Mage_Rose_oral_history (130 kB) Office of NIH History and Stetten Museum Oral History with Dr. Rose Mage, NIAID August 22, 2018 GM: I am Dr. Gordon Margolin, volunteer in the Office of NIH History and Stetten Museum about to do an oral history with Dr. Rose G. Mage. She served as a Career Investigator in the NIAID Lab of Immunology, starting in 1965, and then as the Section Chief of the Molecular Immunogenetics section from 1988 until her retirement in 2008. We are here in the NIH Library’s audio-visual recording center on August 22, 2018. Thank you, Dr. Mage, for agreeing to record this history about you, your scientific accomplishments, and the time you spent here at NIH. Let’s begin by asking you to tell me a bit of your background, where you were raised, something about your family, and your educational pathway. RGM: I was born and raised in New York City. My father was an immigrant from Romania. He arrived in New York when he was 5 years old in 1905. I was named after his mother who died from typhoid in the 1920s. My mother was born in the USA but her family, three of her four older brothers were immigrants. I decided I wanted to be a scientist when I was about 9 years old, did well in public schools and skipped grades twice. I applied to the Bronx High School of Science and was accepted after taking a written test for admission. -

Timeline of Immunology
TIMELINE OF IMMUNOLOGY 1549 – The earliest account of inoculation of smallpox (variolation) occurs in Wan Quan's (1499–1582) 1718 – Smallpox inoculation in Ottoman Empire realized by West. Lady Mary Wortley Montagu, the wife of the British ambassador to Constantinople, observed the positive effects of variolation on the native population and had the technique performed on her own children. 1796 – First demonstration of smallpox vaccination (Edward Jenner) 1837 – Description of the role of microbes in putrefaction and fermentation (Theodore Schwann) 1838 – Confirmation of the role of yeast in fermentation of sugar to alcohol (Charles Cagniard-Latour) 1840 – Proposal of the germ theory of disease (Jakob Henle) 1850 – Demonstration of the contagious nature of puerperal fever (childbed fever) (Ignaz Semmelweis) 1857–1870 – Confirmation of the role of microbes in fermentation (Louis Pasteur) 1862 – Phagocytosis (Ernst Haeckel) 1867 – Aseptic practice in surgery using carbolic acid (Joseph Lister) 1876 – Demonstration that microbes can cause disease-anthrax (Robert Koch) 1877 – Mast cells (Paul Ehrlich) 1878 – Confirmation and popularization of the germ theory of disease (Louis Pasteur) 1880 – 1881 -Theory that bacterial virulence could be attenuated by culture in vitro and used as vaccines. Proposed that live attenuated microbes produced immunity by depleting host of vital trace nutrients. Used to make chicken cholera and anthrax "vaccines" (Louis Pasteur) 1883 – 1905 – Cellular theory of immunity via phagocytosis by macrophages and microphages (polymorhonuclear leukocytes) (Elie Metchnikoff) 1885 – Introduction of concept of a "therapeutic vaccination". Report of a live "attenuated" vaccine for rabies (Louis Pasteur and Pierre Paul Émile Roux). 1888 – Identification of bacterial toxins (diphtheria bacillus) (Pierre Roux and Alexandre Yersin) 1888 – Bactericidal action of blood (George Nuttall) 1890 – Demonstration of antibody activity against diphtheria and tetanus toxins. -

'Asian Nobel' Prizes 18 September 2014
Anti-apartheid hero, ex-Norway PM awarded 'Asian Nobel' prizes 18 September 2014 South African anti-apartheid hero Albie Sachs and Taiwan, were immunologists James P. Allison of the former Norwegian prime minister Gro Harlem MD Anderson Cancer Center at the University of Brundtland, hailed as the "godmother" of Texas, and Tasuku Honjo of Kyoto University for sustainable development, were among five people their contributions in the fight against cancer. Thursday presented with the first Tang Prize, touted as Asia's version of the Nobels. The other recipient was Chinese American historian and Sinologist Yu Ying-shih, the winner in the Taiwan President Ma Ying-jeou presented the Sinology category. awards, which honour outstanding contributions to the environment, human rights, medicine and Each winner received Tw$50 million ($1.7 million), Sinology. with Tw$40 million in cash and the remainder in a grant—a richer purse than the eight million Swedish Brundtland, recognised for her life-long dedication kronor ($1.2 million) that comes with a Nobel Prize. to protection of the environment, called for urgent action to address environmental and climate Named after China's Tang Dynasty (AD 618-907), change over the last quarter of a century. the prize was founded in 2012 with a donation of Tw$3 billion. "We're not far sighted enough to do what is needed" on climate change, she warned, Yin has said he will donate 95 percent of his wealth addressing the guests invited to the presentation to charity during his lifetime. His net assets are ceremony in Taipei. estimated by Forbes magazine at $4.5 billion. -
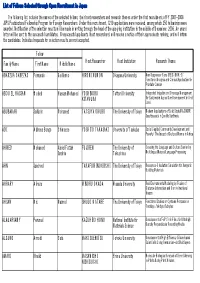
List of Fellows Selected Through Open Recruitment in Japan
List of Fellows Selected through Open Recruitment in Japan The following list includes the names of the selected fellows, their host researchers and research themes under the first recruitment of FY 2007-2008 JSPS Postdoctoral Fellowship Program for Foreign Researchers. Under this recruitment, 1219 applications were received, among which 250 fellowships were awarded. Notification of the selection results will be made in writing through the head of the applying institution in the middle of December, 2006. An award letter will be sent to the successful candidates. Unsuccessful applicants (host researchers) will receive a notice of their approximate ranking, and will inform the candidates. Individual requests for selection results are not accepted. Fellow Family Name First Name Middle Name Host Researcher Host Institution Research Theme ABARZUA CABEZAS Fernando Guillermo HIROMI KUMON Okayama University New Suppressor Gene (REIC/DKK-3): Functional Analysis and Clinical Application for Prostate Cancer ABOU EL HASSAN Waleed Hassan Mohamed YOSHINOBU Tottori University Integrated Irrigation and Drainage Management KITAMURA for Sustainable Agriculture Development in Arid Land ABUBAKAR Saifudin Mohamed TATSUYA OKUBO The University of Tokyo Modern Applications of Solid State MAS NMR Spectroscopy in Zeolite Synthesis ADI Alpheus Bongo Chimaeze YOSHITO TAKASAKI University of Tsukuba Social Capital, Community Development and Poverty: The Impact of Cultural Norms in Africa AHMED Mohamed Abdel Fattah FUJI REN The University of Crossing the Language and Culture Barrier by Ibrahim Tokushima Multilingual Natural Language Processing AHN Jaecheol TAKAFUMI NOGUCHI The University of Tokyo Resources-Circulation Simulation for Inorganic Building Materials AHRARY Alireza MINORU OKADA Waseda University Real Environments Modeling by Fusion of Distance Information and Omni-directional Images AHSAN Md. -

CV Tasuku Honjo
Curriculum Vitae Professor Dr. Tasuku Honjo Name: Tasuku Honjo Born: 27 January 1942 Major Scientific Interest: Mechanism of antibody memory, cancer therapy Academic and Professional Career since 2005 Professor, Department of Immunology and Genomic Medicine, Graduate School of Medicine, Kyoto University, Japan 1984 ‐ 2005 Professor, Department of Medical Chemistry, Faculty of Medicine, Kyoto University, Japan 1979 ‐ 1984 Professor of Genetics, Medical Faculty, Osaka University, Japan 1975 PHD in Medical Chemistry, Kyoto University 1974 Assistant Professor, Medical Faculty, Tokyo University, Japan 1966 MD, Kyoto University Functions in Scientific Societies and Committees (Selection) since 2012 Chairman, Board of Directors, Shizuoka Prefectural University Corporation, Japan 2006 ‐ 2012 Executive Member, Council for Science and Technology Policy, Cabinet Office, Japan since 2006 Scientific advisory board of the Singapore Immunology Network since 2005 Councilor of Takeda Science Foundation 2002 ‐ 2004 Dean, Faculty of Medicine, Kyoto University, Japan Nationale Akademie der Wissenschaften Leopoldina www.leopoldina.org 1 1996 ‐ 2000 Dean, Faculty of Medicine, Kyoto University, Japan since 1996 External advisory board of the Committee for Human Gene Therapy Working Group 1992 ‐ 1995 Fellowship review committee member of International Human Frontier Science Program Honours and Awarded Memberships (Selection) 2018 Nobel Prize in Physiology or Medicine 2016 Kyoto Prize 2012 Robert Koch Prize 2005 Member of Japan Academy 2004 Leading Japanese Scientists -

Recent Winners of the Nobel Medicine Prize 1 October 2018
Recent winners of the Nobel Medicine Prize 1 October 2018 2015: William Campbell (US citizen born in Ireland) and Satoshi Omura (Japan), Tu Youyou (China) for unlocking treatments for malaria and roundworm. 2014: John O'Keefe (Britain, US), Edvard I. Moser and May-Britt Moser (Norway) for discovering how the brain navigates with an "inner GPS". 2013: Thomas C. Suedhof (US citizen born in Germany), James E. Rothman and Randy W. Schekman (US) for work on how the cell organises its transport system. 2012: Shinya Yamanaka (Japan) and John B. Gurdon (Britain) for discoveries showing how adult cells can be transformed back into stem cells. 2011: Bruce Beutler (US), Jules Hoffmann (French citizen born in Luxembourg) and Ralph Steinman (Canada) for work on the body's immune system. Credit: Wikipedia 2010: Robert G. Edwards (Britain) for the development of in-vitro fertilisation. 2009: Elizabeth Blackburn (Australia-US), Carol Here is a list of the winners of the Nobel Medicine Greider and Jack Szostak (US) for discovering how Prize in the past 10 years, after James Allison of chromosomes are protected by telomeres, a key the US and Tasuku Honjo of Japan were awarded factor in the ageing process. Monday for research that has revolutionised cancer treatment: © 2018 AFP 2018: Immunologists Allison and Honjo win for figuring out how to release the immune system's brakes to allow it to attack cancer cells more efficiently. 2017: US geneticists Jeffrey Hall, Michael Rosbash and Michael Young for their discoveries on the internal biological clock that governs the wake- sleep cycles of most living things. -

The24th Keio Medical Science Prize
The 24th Keio Medical Science Prize Prize Laureates 1996 Stanley B. Prusiner, Shigetada Nakanishi 1997 Robert A. Weinberg, Tadatsugu Taniguchi 1998 M. Judah Folkman, Katsuhiko Mikoshiba 1999 Elizabeth H. Blackburn, Shinya Yoshikawa 2000 Arnold J. Levine, Yusuke Nakamura 2001 Tony Hunter, Masatoshi Takeichi 2002 Barry J. Marshall, Koichi Tanaka 2003 Ronald M. Evans, Yasushi Miyashita 2004 Roger Y. Tsien 2005 Yoshinori Fujiyoshi 2006 Thomas A. Steitz 2007 Brian J. Druker, Hiroaki Mitsuya 2008 Fred H. Gage, Shimon Sakaguchi 2009 Jeffrey M. Friedman, Kenji Kangawa 2010 Jules A. Hoffmann, Shizuo Akira 2011 Philip A. Beachy, Keiji Tanaka 2012 Steven A. Rosenberg, Hiroyuki Mano 2013 Victor R. Ambros, Shigekazu Nagata 2014 Karl Deisseroth, Hiroshi Hamada 2015 Jeffrey I. Gordon, Yoshinori Ohsumi 2016 Svante Pääbo, Tasuku Honjo 2017 John E. Dick, Seiji Ogawa 2018 Feng Zhang, Masashi Yanagisawa Deadline: Thursday, March 7, 2019 (Japan local time) Keio University annually awards The Keio Medical Science Prize to recognize researchers who have made an outstanding contribution to the field of medicine and life sciences. Laureates receive a certificate of merit, medal, and a monetary award of 10 million yen. The award ceremony and commemorative lectures are held at Keio University in Tokyo, Japan. 〈Criteria〉 - A researcher who have made a breakthrough in the fields of medicine and life sciences, or a Call related field. - A researcher who have made an outstanding contribution to basic and clinical medicine. 〈Selection〉 13 selection committee members and around 80 specialists from various fields within and outside for Keio University will select laureates through a rigorous review process. 〈Eligibility〉 Nominees must be currently active in their field of research, and be expected to make future contributions to the field. -

IUBMB Newsletter Issue 2.Pdf
IUBMB NEWS Issue 2 October 2016 President’s Message IUBMB aims to promote research and education in In This Issue Biochemistry and Molecular Biology. In this regard, over the President’s Message years we have channelled great efforts into the research aspects of our science. We now consider it timely to turn our Report on the 16th IUBMB attention to educational initiatives. Conference / Young To address this aim and in collaboration with FEBS, we are organising a conference on Education in Biochemistry and Molecular Biology, to be held in Scientists’ Program Rehovot (Israel) in September 2017. The conference seeks to provide a think-tank Tang Prize Foundation setting in which to draw up ideas to improve the current approach to teaching these subjects, and to generate a series of recommendations to be shared with Awardees / IUBMB Travel the educational community. In this regard, this conference will serve to propel the Fellowships changes that are required to bring education in biochemistry and molecular biology st at all levels into the 21 century. Another Way to Think About Another important aspect of education is to provide young scientists from Disease by Gregory Petsko developing countries the opportunity to attend international congresses and meetings. IUBMB is firmly committed to this endeavour and devotes significant Retrospective — financial resources to this end. Given that the number of applications from African countries is very low, I wish to encourage our young African colleagues to apply Osamu Hayaishi and benefit from IUBMB travel awards and Wood-Whelan fellowships. An Appreciation — IUBMB is also launching a new activity, namely Focused Meetings. -

Tasuku Honjo Kyoto University, Kyoto, Japan
333 Serendipities of Acquired Immunity Nobel Lecture, December 7, 2018 by Tasuku Honjo Kyoto University, Kyoto, Japan. INTRODUCTION For a long time, biology was perceived as the lesser of the natural sciences because, unlike physics, deductive reasoning could not be used to solve biological problems. Biology has been full of mystery since I started my career in biological sciences almost half a century ago. Although the basic principles of biology stem from the rules of physics, biological systems have such an extraordinary, layered complexity derived from a tremen- dous number of parameters, beautifully and magically intertwined and controlled to achieve what we call “life”. Paradoxically, we start with a rather limited number (about 20,000) of coding region genes. However, many transcripts can be generated from a single coding gene locus, indi- cating that a single gene can produce many proteins. Furthermore, there are a much higher number of non-coding transcripts that may afect the expression of the coding genes. DNA, RNA and proteins can be chemically modifed by methylation, phosphorylation and acetylation. In addition, at least 20,000 metabolites circulate in our blood. These can also be sensed by cells, interacting with various proteins and infuencing gene expres- sion, thus generating enormously complicated regulatory mechanisms to achieve homeostasis. The origin of metabolites can be traced not only to the biochemistry of our own cells but also to the diverse communities of 334 THE NOBEL PRIZES microbes inhabiting every surface of the body. If we imagine roughly 1013 order of our own cells, each expressing diferent proteins and containing diferent metabolites, in constant dialog with 1014 order of microbial cells, also in diferent metabolic states, the complexity of our biological system exceeds by far the physical and chemical complexity of the universe.