A Transcriptomics-Based Meta-Analysis Combined
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -
Primepcr™Assay Validation Report
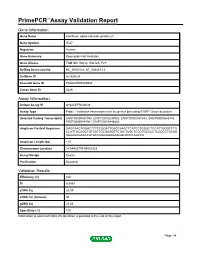
PrimePCR™Assay Validation Report Gene Information Gene Name interferon, alpha-inducible protein 27 Gene Symbol IFI27 Organism Human Gene Summary Description Not Available Gene Aliases FAM14D, ISG12, ISG12A, P27 RefSeq Accession No. NC_000014.8, NT_026437.12 UniGene ID Hs.532634 Ensembl Gene ID ENSG00000165949 Entrez Gene ID 3429 Assay Information Unique Assay ID qHsaCEP0024638 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000557098, ENST00000298902, ENST00000557634, ENST00000555744, ENST00000444961, ENST00000448882 Amplicon Context Sequence GAGCAACTGGACTCTCCGGATTGACCAAGTTCATCCTGGGCTCCATTGGGTCTG CCATTGCGGCTGTCATTGCGAGGTTCTACTAGCTCCCTGCCCCTCGCCCTGCAG AGAAGAGAACCATGCCAGGGGAGAAGGCACCCAGCCA Amplicon Length (bp) 115 Chromosome Location 14:94582779-94582923 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 23.09 cDNA Tm (Celsius) 86 gDNA Cq 23.06 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report IFI27, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus
Gao et al. Hereditas (2021) 158:3 https://doi.org/10.1186/s41065-020-00167-5 RESEARCH Open Access IFI27 may predict and evaluate the severity of respiratory syncytial virus infection in preterm infants Junyan Gao1, Xueping Zhu2, Mingfu Wu1, Lijun Jiang1, Fudong Wang1 and Shan He2,3* Abstract Background: Preterm infants are a special population that vulnerable to respiratory syncytial virus (RSV) infection and the lower respiratory tract infections (LRTIs) caused by RSV could be severe and even life-threating. The purpose of the present study was to identify candidate genes of preterm infants who are susceptible to RSV infection and provide a new insight into the pathogenesis of RSV infection. Methods: Three datasets (GSE77087, GSE69606 and GSE41374) containing 183 blood samples of RSV infected patients and 33 blood samples of healthy controls from Gene Expression Omnibus (GEO) database were downloaded and the differentially expressed genes (DEGs) were screened out. The function and pathway enrichments were analyzed through Database for Annotation, Visualization and Integrated Discovery (DAVID) website. The protein-protein interaction (PPI) network for DEGs was constructed through Search Tool for the Retrieval of Interacting Genes (STRING). The module analysis was performed by Cytoscape software and hub genes were identified. Clinical verification was employed to verify the expression level of top five hub genes among 72 infants including 50 RSV infected patients and 22 non-RSV-infected patients hospitalized in our center. Further, the RSV infected infants with high-expression IFI27 and those with low-expression IFI27 were compared (defined as higher or lower than the median mRNA level). -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus Infection in Preterm Infants
IFI27 May Predict and Evaluate the Severity of Respiratory Syncytial Virus Infection in Preterm Infants Junyan Gao Aliated Hospital of Yangzhou University https://orcid.org/0000-0001-6678-6147 Xueping Zhu Children's Hospital of Soochow University Mingfu Wu Aliated Hospital of Yangzhou University Lijun Jiang Aliated Hospital of Yangzhou University Fudong Wang Aliated Hospital of Yangzhou University Shan He ( [email protected] ) Research Keywords: respiratory syncytial virus, preterm infants, differentially expressed genes, protein‐protein interaction, pathway enrichment, interferon alpha inducible protein 27 Posted Date: July 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-42880/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on January 2nd, 2021. See the published version at https://doi.org/10.1186/s41065-020-00167-5. Page 1/21 Abstract Background: Preterm infants are a special population that vulnerable to respiratory syncytial virus (RSV) infection and the lower respiratory tract infection (LRTIs) caused by RSV could be severe and even life- threating. The purpose of the present study was to identify candidate genes of preterm infants who are susceptible to RSV infection and provide a new insight into the pathogenesis of RSV infection. Methods: Three datasets (GSE77087, GSE69606 and GSE41374) containing 183 blood samples of RSV infected patients and 33 blood samples of healthy controls from Gene Expression Omnibus (GEO) database were downloaded and the differentially expressed genes (DEGs) were screened out. The function and pathway enrichments were analyzed through Database for Annotation, Visualization and Integrated Discovery (DAVID) website. -

Microrna-Mediated Networks Underlie Immune Response Regulation in Papillary Thyroid 51
OPEN MicroRNA-mediated networks underlie SUBJECT AREAS: immune response regulation in papillary GENE REGULATORY NETWORKS thyroid carcinoma CANCER GENOMICS Chen-Tsung Huang1, Yen-Jen Oyang1, Hsuan-Cheng Huang4 & Hsueh-Fen Juan1,2,3 Received 1 2 15 July 2014 Graduate Institute of Biomedical Electronics and Bioinformatics, National Taiwan University, Taipei, Taiwan, Department of Life Science, National Taiwan University, Taipei, Taiwan, 3Institute of Molecular and Cellular Biology, National Taiwan University, Accepted Taipei, Taiwan, 4Institute of Biomedical Informatics and Center for Systems and Synthetic Biology, National Yang-Ming University, 9 September 2014 Taipei, Taiwan. Published 29 September 2014 Papillary thyroid carcinoma (PTC) is a common endocrine malignancy with low death rate but increased incidence and recurrence in recent years. MicroRNAs (miRNAs) are small non-coding RNAs with diverse regulatory capacities in eukaryotes and have been frequently implied in human cancer. Despite current Correspondence and progress, however, a panoramic overview concerning miRNA regulatory networks in PTC is still lacking. requests for materials Here, we analyzed the expression datasets of PTC from The Cancer Genome Atlas (TCGA) Data Portal and demonstrate for the first time that immune responses are significantly enriched and under specific should be addressed to regulation in the direct miRNA2target network among distinctive PTC variants to different extents. H.C.H. (hsuancheng@ Additionally, considering the unconventional properties of miRNAs, we explore the protein-coding ym.edu.tw) or H.F.J. competing endogenous RNA (ceRNA) and the modulatory networks in PTC and unexpectedly disclose ([email protected]) concerted regulation of immune responses from these networks. Interestingly, miRNAs from these conventional and unconventional networks share general similarities and differences but tend to be disparate as regulatory activities increase, coordinately tuning the immune responses that in part account for PTC tumor biology. -

A De Novo Missense Mutation of FGFR2 Causes Facial Dysplasia Syndrome in Holstein Cattle Jørgen S
Agerholm et al. BMC Genetics (2017) 18:74 DOI 10.1186/s12863-017-0541-3 RESEARCH ARTICLE Open Access A de novo missense mutation of FGFR2 causes facial dysplasia syndrome in Holstein cattle Jørgen S. Agerholm1*, Fintan J. McEvoy1, Steffen Heegaard2,3, Carole Charlier4, Vidhya Jagannathan5 and Cord Drögemüller5 Abstract Background: Surveillance for bovine genetic diseases in Denmark identified a hitherto unreported congenital syndrome occurring among progeny of a Holstein sire used for artificial breeding. A genetic aetiology due to a dominant inheritance with incomplete penetrance or a mosaic germline mutation was suspected as all recorded cases were progeny of the same sire. Detailed investigations were performed to characterize the syndrome and to reveal its cause. Results: Seven malformed calves were submitted examination. All cases shared a common morphology with the most striking lesions being severe facial dysplasia and complete prolapse of the eyes. Consequently the syndrome was named facial dysplasia syndrome (FDS). Furthermore, extensive brain malformations, including microencephaly, hydrocephalus, lobation of the cerebral hemispheres and compression of the brain were present. Subsequent data analysis of progeny of the sire revealed that around 0.5% of his offspring suffered from FDS. High density single nucleotide polymorphism (SNP) genotyping data of the seven cases and their parents were used to map the defect in the bovine genome. Significant genetic linkage was obtained for three regions, including chromosome 26 where whole genome sequencing of a case-parent trio revealed two de novo variants perfectly associated with the disease: an intronic SNP in the DMBT1 gene and a single non-synonymous variant in the FGFR2 gene. -

Transcriptional Recapitulation and Subversion Of
Open Access Research2007KaiseretVolume al. 8, Issue 7, Article R131 Transcriptional recapitulation and subversion of embryonic colon comment development by mouse colon tumor models and human colon cancer Sergio Kaiser¤*, Young-Kyu Park¤†, Jeffrey L Franklin†, Richard B Halberg‡, Ming Yu§, Walter J Jessen*, Johannes Freudenberg*, Xiaodi Chen‡, Kevin Haigis¶, Anil G Jegga*, Sue Kong*, Bhuvaneswari Sakthivel*, Huan Xu*, Timothy Reichling¥, Mohammad Azhar#, Gregory P Boivin**, reviews Reade B Roberts§, Anika C Bissahoyo§, Fausto Gonzales††, Greg C Bloom††, Steven Eschrich††, Scott L Carter‡‡, Jeremy E Aronow*, John Kleimeyer*, Michael Kleimeyer*, Vivek Ramaswamy*, Stephen H Settle†, Braden Boone†, Shawn Levy†, Jonathan M Graff§§, Thomas Doetschman#, Joanna Groden¥, William F Dove‡, David W Threadgill§, Timothy J Yeatman††, reports Robert J Coffey Jr† and Bruce J Aronow* Addresses: *Biomedical Informatics, Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229, USA. †Departments of Medicine, and Cell and Developmental Biology, Vanderbilt University and Department of Veterans Affairs Medical Center, Nashville, TN 37232, USA. ‡McArdle Laboratory for Cancer Research, University of Wisconsin, Madison, WI 53706, USA. §Department of Genetics and Lineberger Cancer Center, University of North Carolina, Chapel Hill, NC 27599, USA. ¶Molecular Pathology Unit and Center for Cancer Research, Massachusetts deposited research General Hospital, Charlestown, MA 02129, USA. ¥Division of Human Cancer Genetics, The Ohio State University College of Medicine, Columbus, Ohio 43210-2207, USA. #Institute for Collaborative BioResearch, University of Arizona, Tucson, AZ 85721-0036, USA. **University of Cincinnati, Department of Pathology and Laboratory Medicine, Cincinnati, OH 45267, USA. ††H Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA. ‡‡Children's Hospital Informatics Program at the Harvard-MIT Division of Health Sciences and Technology (CHIP@HST), Harvard Medical School, Boston, Massachusetts 02115, USA. -

Identification of Key Genes and Pathways in Pancreatic Cancer
G C A T T A C G G C A T genes Article Identification of Key Genes and Pathways in Pancreatic Cancer Gene Expression Profile by Integrative Analysis Wenzong Lu * , Ning Li and Fuyuan Liao Department of Biomedical Engineering, College of Electronic and Information Engineering, Xi’an Technological University, Xi’an 710021, China * Correspondence: [email protected]; Tel.: +86-29-86173358 Received: 6 July 2019; Accepted: 7 August 2019; Published: 13 August 2019 Abstract: Background: Pancreatic cancer is one of the malignant tumors that threaten human health. Methods: The gene expression profiles of GSE15471, GSE19650, GSE32676 and GSE71989 were downloaded from the gene expression omnibus database including pancreatic cancer and normal samples. The differentially expressed genes between the two types of samples were identified with the Limma package using R language. The gene ontology functional and pathway enrichment analyses of differentially-expressed genes were performed by the DAVID software followed by the construction of a protein–protein interaction network. Hub gene identification was performed by the plug-in cytoHubba in cytoscape software, and the reliability and survival analysis of hub genes was carried out in The Cancer Genome Atlas gene expression data. Results: The 138 differentially expressed genes were significantly enriched in biological processes including cell migration, cell adhesion and several pathways, mainly associated with extracellular matrix-receptor interaction and focal adhesion pathway in pancreatic cancer. The top hub genes, namely thrombospondin 1, DNA topoisomerase II alpha, syndecan 1, maternal embryonic leucine zipper kinase and proto-oncogene receptor tyrosine kinase Met were identified from the protein–protein interaction network. -

Identifying Robust Biomarkers of Infection Through an Omics-Based
medRxiv preprint doi: https://doi.org/10.1101/2020.07.28.20163329; this version posted July 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . 1 Identifying robust biomarkers of infection 2 through an omics-based meta-analysis 3 4 Ashleigh C Myall1,2, Simon Perkins1, David Rushton4, Jonathan David5, Phillippa Spencer6, 5 Andrew R Jones1,&, & Philipp Antczak1,3,&* 6 1Institute of Systems, Molecular and Integrative Biology, University of Liverpool, Liverpool, 7 United Kingdom 8 2Department of Mathematics, Imperial College London, London, United Kingdom 9 3Center for Molecular Medicine, University of Cologne, Cologne, Germany 10 4Defence and Security Analysis Division, Defence Science and Technology laboratory 11 (DSTL), Porton Down Salisbury, United Kingdom 12 5Chemical, Biological and Radiological Division, Defence Science and Technology 13 laboratory (DSTL), Porton Down Salisbury, United Kingdom 14 6Cyber and Information Systems Division, Defence Science and Technology laboratory 15 (DSTL), Porton Down, Salisbury, United Kingdom 16 17 *Corresponding author 18 E-mail: [email protected] (PA) 19 &Joint Senior Authors 1 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2020.07.28.20163329; this version posted July 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. -

Mouse Ifi27l2a Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Ifi27l2a Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Ifi27l2a conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Ifi27l2a gene (NCBI Reference Sequence: NM_029803 ; Ensembl: ENSMUSG00000079017 ) is located on Mouse chromosome 12. 4 exons are identified, with the ATG start codon in exon 1 and the TGA stop codon in exon 4 (Transcript: ENSMUST00000055071). Exon 3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Ifi27l2a gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP24-62E20 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a knock-out allele exhibit normal susceptibility to infection with H1N1 or H7N7 influenza A virus, although sloughing of dead cells into bronchiolar lumen persists somewhat longer after infection than in infected controls. Exon 3 is not frameshift exon, and covers 60.0% of the coding region. The size of intron 2 for 5'-loxP site insertion: 488 bp, and the size of intron 3 for 3'-loxP site insertion: 448 bp. The size of effective cKO region: ~630 bp. The cKO region does not have any other known gene. Page 1 of 7 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 3 4 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Homology arm Exon of mouse Ifi27l2a cKO region loxP site Page 2 of 7 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Supplementary Data
Supplemental figures Supplemental figure 1: Tumor sample selection. A total of 98 thymic tumor specimens were stored in Memorial Sloan-Kettering Cancer Center tumor banks during the study period. 64 cases corresponded to previously untreated tumors, which were resected upfront after diagnosis. Adjuvant treatment was delivered in 7 patients (radiotherapy in 4 cases, cyclophosphamide- doxorubicin-vincristine (CAV) chemotherapy in 3 cases). 34 tumors were resected after induction treatment, consisting of chemotherapy in 16 patients (cyclophosphamide-doxorubicin- cisplatin (CAP) in 11 cases, cisplatin-etoposide (PE) in 3 cases, cisplatin-etoposide-ifosfamide (VIP) in 1 case, and cisplatin-docetaxel in 1 case), in radiotherapy (45 Gy) in 1 patient, and in sequential chemoradiation (CAP followed by a 45 Gy-radiotherapy) in 1 patient. Among these 34 patients, 6 received adjuvant radiotherapy. 1 Supplemental Figure 2: Amino acid alignments of KIT H697 in the human protein and related orthologs, using (A) the Homologene database (exons 14 and 15), and (B) the UCSC Genome Browser database (exon 14). Residue H697 is highlighted with red boxes. Both alignments indicate that residue H697 is highly conserved. 2 Supplemental Figure 3: Direct comparison of the genomic profiles of thymic squamous cell carcinomas (n=7) and lung primary squamous cell carcinomas (n=6). (A) Unsupervised clustering analysis. Gains are indicated in red, and losses in green, by genomic position along the 22 chromosomes. (B) Genomic profiles and recurrent copy number alterations in thymic carcinomas and lung squamous cell carcinomas. Gains are indicated in red, and losses in blue. 3 Supplemental Methods Mutational profiling The exonic regions of interest (NCBI Human Genome Build 36.1) were broken into amplicons of 500 bp or less, and specific primers were designed using Primer 3 (on the World Wide Web for general users and for biologist programmers (see Supplemental Table 2) [1].