207534Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evolution and Understanding of the D-Block Elements in the Periodic Table Cite This: Dalton Trans., 2019, 48, 9408 Edwin C
Dalton Transactions View Article Online PERSPECTIVE View Journal | View Issue Evolution and understanding of the d-block elements in the periodic table Cite this: Dalton Trans., 2019, 48, 9408 Edwin C. Constable Received 20th February 2019, The d-block elements have played an essential role in the development of our present understanding of Accepted 6th March 2019 chemistry and in the evolution of the periodic table. On the occasion of the sesquicentenniel of the dis- DOI: 10.1039/c9dt00765b covery of the periodic table by Mendeleev, it is appropriate to look at how these metals have influenced rsc.li/dalton our understanding of periodicity and the relationships between elements. Introduction and periodic tables concerning objects as diverse as fruit, veg- etables, beer, cartoon characters, and superheroes abound in In the year 2019 we celebrate the sesquicentennial of the publi- our connected world.7 Creative Commons Attribution-NonCommercial 3.0 Unported Licence. cation of the first modern form of the periodic table by In the commonly encountered medium or long forms of Mendeleev (alternatively transliterated as Mendelejew, the periodic table, the central portion is occupied by the Mendelejeff, Mendeléeff, and Mendeléyev from the Cyrillic d-block elements, commonly known as the transition elements ).1 The periodic table lies at the core of our under- or transition metals. These elements have played a critical rôle standing of the properties of, and the relationships between, in our understanding of modern chemistry and have proved to the 118 elements currently known (Fig. 1).2 A chemist can look be the touchstones for many theories of valence and bonding. -

Historical Development of the Periodic Classification of the Chemical Elements
THE HISTORICAL DEVELOPMENT OF THE PERIODIC CLASSIFICATION OF THE CHEMICAL ELEMENTS by RONALD LEE FFISTER B. S., Kansas State University, 1962 A MASTER'S REPORT submitted in partial fulfillment of the requirements for the degree FASTER OF SCIENCE Department of Physical Science KANSAS STATE UNIVERSITY Manhattan, Kansas 196A Approved by: Major PrafeLoor ii |c/ TABLE OF CONTENTS t<y THE PROBLEM AND DEFINITION 0? TEH-IS USED 1 The Problem 1 Statement of the Problem 1 Importance of the Study 1 Definition of Terms Used 2 Atomic Number 2 Atomic Weight 2 Element 2 Periodic Classification 2 Periodic Lav • • 3 BRIEF RtiVJiM OF THE LITERATURE 3 Books .3 Other References. .A BACKGROUND HISTORY A Purpose A Early Attempts at Classification A Early "Elements" A Attempts by Aristotle 6 Other Attempts 7 DOBEREBIER'S TRIADS AND SUBSEQUENT INVESTIGATIONS. 8 The Triad Theory of Dobereiner 10 Investigations by Others. ... .10 Dumas 10 Pettehkofer 10 Odling 11 iii TEE TELLURIC EELIX OF DE CHANCOURTOIS H Development of the Telluric Helix 11 Acceptance of the Helix 12 NEWLANDS' LAW OF THE OCTAVES 12 Newlands' Chemical Background 12 The Law of the Octaves. .........' 13 Acceptance and Significance of Newlands' Work 15 THE CONTRIBUTIONS OF LOTHAR MEYER ' 16 Chemical Background of Meyer 16 Lothar Meyer's Arrangement of the Elements. 17 THE WORK OF MENDELEEV AND ITS CONSEQUENCES 19 Mendeleev's Scientific Background .19 Development of the Periodic Law . .19 Significance of Mendeleev's Table 21 Atomic Weight Corrections. 21 Prediction of Hew Elements . .22 Influence -

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

The Rare Earths II
Redis co very of the Elements The Ra re Earth s–The Con fusing Years I A gallery of rare earth scientists and a timeline of their research I I James L. Marshall, Beta Eta 1971 , and Virginia R. Marshall, Beta Eta 2003 , Department of Chemistry, University of North Texas, Denton, TX 76203-5070, [email protected] The rare earths after Mosander. In the pre - vi ou s HEXAGON “Rediscovery” article, 1p we were introduced to the 17 rare earths, found in the f-block and the Group III chemical family of Figure 1. Important scientists dealing with rare earths through the nineteenth century. Johan Gadolin the Periodic Table. Because of a common (1760 –1852) 1g —discovered yttrium (1794). Jöns Jacob Berzelius (1779 –1848) and Martin Heinrich valence electron configuration, the rare earths Klaproth (1743 –1817) 1d —discovered cerium (1803). Carl Gustaf Mosander (1787 –1858) 1p —discovered have similar chemical properties, and their lanthanum (1839), didymium (1840), terbium, and erbium (1843). Jean-Charles deGalissard Marignac chemical separation from one another can be (1817 –1894) 1o —discovered ytterbium (1878) and gadolinium (1880). Per Teodor Cleve (1840 –1905) 1n — difficult. From preparations of the first two rare discovered holmium and thulium (1879). Lars Fredrik Nilson (1840 –1899) 1n —discovered scandium earth element s—yttrium and ceriu m—the (1879). Paul-Émile Lecoq de Boisbaudran (1838 –1912) —discovered samarium (1879) and dysprosium Swedish chemist Carl Gustaf Mosander (Figure (1886). 1b Carl Auer von Welsbach (1858 –1929) 1c —discovered praseodymium and neodymium (1885); 1, 2) was able to separate four additional ele - co-discovered lutetium (1907). -

Astrid Cleve Von Euler Född 11 Januari 1875, Död 8 April 1968, Dotter Till Professor (Uppsala) Per Teodor Cleve Och Författaren Caralma Öhbom
Sveriges första kvinnliga doktor i naturvetenskapliga ämnen var lärare på Norra Real 1912-16 Astrid Cleve von Euler född 11 januari 1875, död 8 april 1968, dotter till professor (Uppsala) Per Teodor Cleve och författaren Caralma Öhbom. Disputerade i botanik den 27 maj 1898 vid Uppsala universitet. Astrid Cleve utbildade sig till botanist och blev fil. kand. 1894 och disputerade i Uppsala 1898. Hon var därmed den tredje kvinnan i Sverige som disputerade. (Ellen Fries var den första kvinnan som doktorerade, 1883 i historia). Hon var starkt målinriktad på att bli forskare men när hon som kvinna inte accepterades som sådan, blev hon lärare istället. Hon fick mycket goda betyg i undervisningsskicklighet och därmed tjänst som lärare på Norra Real. Naturligtvis inte som lektor på gymnasiet - kvinnor kunde inte få sådan tjänst - men genom sin kompetens kom hon ändå att vikariera som lektor med en lön som så småningom blev 75,9 procent av vad hennes manliga kolleger fick. Lönefrågan diskuterades i många instanser innan den här procentsatsen fastställdes. Man var helt enkelt rädd att statuera exempel. ”... doktor Cleve von Euler gjorda framställningen knappast lärer kunna anses vara prejudicerande i avseende å kvinnors anställning i statstjänst. Ett dylikt bifall kommer framför allt ej att beröra det mycket omstridda spörsmålet om kvinnas lämplighet eller icke såsom undervisare för manliga lärjungar å högre stadium, ...” Astrid Cleve gifte sig den 2 september 1902 med Hans von Euler, som 1929 skulle få nobelpriset i kemi. Han var invandrare från Tyskland, men hade då bott i Sverige tillräckligt länge - och var av ”god frejd” - för att få gifta sig här. -

Carl Wilhelm Scheele (1742-1786) in the Literature Between the 19Th and 21Th Centuries
Revista CENIC. Ciencias Químicas ISSN: 1015-8553 ISSN: 2221-2442 [email protected] Centro Nacional de Investigaciones Científicas Cuba Immortal fame of the Swedish apothecary and chemist: Carl Wilhelm Scheele (1742-1786) in the literature between the 19th and 21th centuries Sztejnberg, Aleksander Immortal fame of the Swedish apothecary and chemist: Carl Wilhelm Scheele (1742-1786) in the literature between the 19th and 21th centuries Revista CENIC. Ciencias Químicas, vol. 49, no. 1, 2018 Centro Nacional de Investigaciones Científicas, Cuba Available in: https://www.redalyc.org/articulo.oa?id=181661081016 PDF generated from XML JATS4R by Redalyc Project academic non-profit, developed under the open access initiative Aleksander Sztejnberg. Immortal fame of the Swedish apothecary and chemist: Carl Wilhelm Scheele (... Articulos de Revision Immortal fame of the Swedish apothecary and chemist: Carl Wilhelm Scheele (1742-1786) in the literature between the 19th and 21th centuries Fama inmortal del boticario y químico sueco Carl Wilhelm Scheele (1742-1786) en los siglos XIX y XXI Aleksander Sztejnberg a Redalyc: https://www.redalyc.org/articulo.oa? University of Opole, Polonia id=181661081016 [email protected] Received: 02 August 2018 Accepted: 04 September 2018 Abstract: In this article the literature on Carl Wilhelm Scheele (1742-1786) is reviewed, including books on chemistry and the history of chemistry, published between the 18th and the 21st century in different countries in order to: 1) familiarize readers with the names of authors of biographies and biographical notes about Carl Wilhelm Scheele, published in books in 1833-2017, 2) discusse the priority problem for the discovery of oxygen by Joseph Priestley (1733-1804) and Scheele, 3) familiarize readers with literature sources regarding Scheele's letter to Antoine Laurent Lavoisier (1743-1794), written on September 30, 1774, 4) show the causes of Scheele’s premature death, 5) familiarize readers with opinions about Scheele expressed by chemists in 1819-1942. -

204200Orig1s000 204200Orig2s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 204200Orig1s000 204200Orig2s000 MEDICAL REVIEW(S) CLINICAL REVIEW Application Type NDA Application Number(s) 204-200 and 204-640 Priority or Standard Standard Submit Date(s) March 7, 2012 Received Date(s) March 7, 2012 PDUFA Goal Date January 7, 2013 Division / Office DPARP/OND Reviewer Name(s) Peter Starke, MD Review Completion Date October 29, 2012 Established Name Epinephrine injection, USP, 1mg/mL (Proposed) Trade Name Adrenalin® Therapeutic Class Catecholamine Applicant JHP Pharmaceuticals, LLC Formulation(s) Solution for injection Proposed Dosing Hypersensitivity reactions: IM or SC Regimen injection(b) (4) [Ophthalmic use: Topical irrigation or intraocular bolus injection] Proposed Indication(s) Hypersensitivity reactions: severe acute anaphylactic reactions, (b) (4) [Ophthalmic use: induction and maintenance of mydriasis during cataract surgery] Intended Population(s) Hypersensitivity reactions: No age restrictions [Ophthalmic use: No age restrictions] Reference ID: 3209828 Clinical Review ● Peter Starke, MD NDA 204-200 ● Adrenalin® (epinephrine) MEDICAL OFFICER REVIEW Division of Pulmonary, Allergy and Rheumatology Products (DPARP) SUBMISSIONS REVIEWED IN THIS DOCUMENT Document Date CDER Stamp Date Submission Comments March 7, 2012 March 7, 2012 SD-1, eCTD-0000 New NDA submission April 9, 2012 April 9, 2012 SD-2, eCTD-0001 PREA waiver request – Anaphylaxis April 9, 2012 April 9, 2012 SD-3, eCTD-0002 PREA waiver request – Mydriasis April 18, 2012 April 18, 2012 SD-4, eCTD-0003 Request for proprietary name review Sept 5, 2012 Sept 6, 2012 SD-22, eCTD-0021 Draft Ophthalmic Labeling Oct 22, 2012 Oct 22, 2012 SD-27, eCDT-0026 Certification that EpiPen is the reference for the 505(b)(2) application RECOMMENDED REGULATORY ACTION NDA/SUPPLEMENTS: X APPROVAL COMPLETE RESPONSE OTHER ACTION: 2 Reference ID: 3209828 Clinical Review ● Peter Starke, MD NDA 204-200 ● Adrenalin® (epinephrine) Table of Contents 1 RECOMMENDATIONS/RISK BENEFIT ASSESSMENT ........................................ -

Scandium Fathi Habashi
Laval University From the SelectedWorks of Fathi Habashi 2008 Scandium Fathi Habashi Available at: https://works.bepress.com/fathi_habashi/743/ Metall-Magazin Scandium Habashi, F. (1) When the Russian chemist Dmitri Ivanovich num. Although scan- that he called thortveitite, contained Mendeleev (1834-1907) proposed the Periodic dium has two elec- 30 to 40% of Sc2O3. This mineral was Table in 1869 he predicted the existence of trons in the outermost also found later, in Madagascar. It has eka-boron, which would have an atomic weight shell and one would the composition (Sc,Y)2Si2O7. between 40 of calcium and 48 of titanium. The expect that it would Bazzite: Be3(Sc,Al)2Si6O18 is another element was discovered by Swedish chemist Lars form divalent com- silicate mineral containing scandium Fredrik Nilson (1840-1899) in 1878 by spec- pounds, yet its most discovered in 1915. troscopic analysis of the minerals euxenite and common valency state Euxenite: (Y,Ca,Ce,U,Th)(Nb,Ta,Ti)2O6 gadolinite, which had not yet been found any- is three resembling occurs in granite pegmatites and detri- where except in Scandinavia. By processing 10 aluminum. tal black sands. It is commonly par- kg of euxenite and other residues of rare-earth Scandium is usually tially amorphous due to radiation associated with the damage.It was first described in 1870 minerals, Nilson was able to prepare about 2 lanthanides and the and named from the Greek word g of the oxide of the new metal in high purity metal has hexago- meaning hospitable or friendly to and called the metal scandium in honour of his nal structure like the strangers, in allusion to the many rare fatherland. -

Nov. Vortex 2019Vs 3 .Indd
AMERICAN CHEMICAL SOCIETY CALIFORNIA SECTION t Organization t VOLUME LXXXI NUMBER 9 NOVEMBER 2019 Non-Profi U.S. POSTAGE U.S. TIME VALUE CALIFORNIA SECTION CALIFORNIA AMERICAN CHEMICAL SOCIETY CHEMICAL AMERICAN PLEASE DO NOT DELAY - DATED NOTICE INSIDE NOTICE - DATED DO DELAY PLEASE NOT SEE REPORT ON PAGE 4 TABLE OF CONTENTS NOVEMBER MEETING PAGE 2 CHAIR'S MESSAGE PAGE 3 WCC MEETING REPORT (N. DAVIS) PAGE 4 IT'S ELEMENTARY PART 3 (B. MOTZER) PAGE 5 NATURAL GMO DEBUNKED (GMWATCH) PAGE 7 BUSINESS DIRECTORY PAGE 11 INDEX OF ADVERTISERS PAGE 11 BUSINESS DIRECTORY California Section AI and Chemistry: Protein Engineering and East Bay Biotech Tuesday – November 19, 2019 – 6:00 to 9:00 PM Amyris 5885 Hollis St. Suite 100 Emeryville, CA 94608 6:00 PM Networking (Refreshments provided) 7:00 PM Welcome, Panel, and Q&A 7:45 PM Concluding Remarks 7:50 PM Networking and Refreshments Discussion on East Bay research - Come join us to learn more about the future of chemistry, protein engineering, and artificial intelligence within the biotech industry. INDEXINDEX OFOF ADVERTISERSADVERTISERS RobertsonRobertson MiMicrolitcrolit BBPP RSVP here! Guests will sign a non-disclosure upon sign-in at the event. Event access is through the general access door facing Hollis Street. Our Distinguished Panelists: Yue Yang, PhD Loren Perelman, PhD Louis Metzger, PhD Amyris Riffyn Tierra Biosciences Director, Program Vice President, Scientific Chief Scientific Officer Management Solutions The event is FREE and open to the community. More information at: calacs.org or email [email protected] NOVEMBER 2019 P AGE 2 PAGE 11 THE VORTEX has been known to occur for decades and most forty years. -

Holmium Same As Santa’S Signature Hearty Laugh
Ho Ho Ho, Merry Christmas! It’s that time of the year, it’s Christmas time! Council of the 118 Elements I’m thrilled to introduce you to the element who has a chemical symbol the Santa Claus ~ Ho…Ho…Holmium same as Santa’s signature hearty laugh. Let’s give a warm welcome to Holmium in this chilly month of Christmas. Name Holmium He is a member of the Lanthanide series. The Lanthanides are put in a Atomic No. Atomic Weight disembodied block below the main body of the Periodic Table. It comprises 67 164.9303 15 elements with atomic numbers 57 to 71, from Lanthanum to Lutetium. Origin of the Name Home town of Cleve Holmia ‘Stockholm’ (the discoverer) Holmium has the highest magnetic strength of any element, and therefore is used to create the strongest artificially generated magnetic Melting Point 1461 °C Electronic fields. Another special trait is that he readily absorbs excess neutrons Arrangement Boiling Point 2720 °C 2,8,18,29,8,2 and so controls the chain reaction that fuels the nuclear reactor. Density 8.54 g/cm3 Humans also owe a huge thanks to Holmium for his Abundance 1.3 mg/kg significant contributions in the medical and dental fields. Category Metal Holmium lasers can produce a wavelength of light close to that of a microwave oven. Such electromagnetic radiation Discovery of the is efficiently absorbed by water molecules because it Santa Element Discoverer : Swede perfectly excites the hydrogen-oxygen bonds in water. Per Teodor Cleve Soft tissue in our bodies is largely made up of water and Year of Discovery: 1878 Cleve removed all of the known these lasers are energetic enough to cut through flesh. -

Kemisk Tidskrift I Tiden
tidningen begynnelsen var Notiser. Kemiska Sällskapet i Stockholm grundades Kemisk den 22 november 1883. Den 10 sep- tember 1885 ändrade man namnet I till Kemistsamfundet. Det fanns ingen Tidskrift i tiden självständigt facktidskrift för ren och till- lämpad kemi på svenska. [Av Ulf Ulfvarson, professor emeritus, KTH] Teknisk Tidskrift, utgiven av Svenska Teknologföreningen sedan 1871, var för August Strindberg läste den, berömda kemister bred. Förhandlingar mellan Kemistsam- fundet och Svenska Teknologföreningen skrev artiklar i den. En period drog den in stora om att ge ut en kemitidning var resul- tatlösa. pengar på annonser. Nu är tidningens framtid rätt Den 21 april 1887 beslöt Kemistsamfun- det att på prov ge ut Kemiska Notiser. Ut- oviss, skriver Ulf Ulfvarson. givningen fortsatte i två år. Redan denna lilla publikation blev en succé att döma av intresset att medverka hos andra kemist- sammanslutningar. Även lekmän tycks ha läst den. Författaren August Strindberg, känd som hobbykemist, hade i sin kvar- låtenskap exemplar av Kemiska Notiser. Svensk Kemisk Tidskrift. Uppmuntrade av framgången ändrade Kemistsamfun- det år 1989 namnet på publikationen till Svensk Kemisk Tidskrift, sida t v. Under några år fanns Kemiska Notiser kvar som undertitel (utom i de första numren). Fler huvudmän tillkom med tiden: kemistsam- manslutningarna i Uppsala och Lund, Ke- miska sällskapet i Stockholm (tidigare Ke- mistföreningen vid Stockholms högskola), Kemisk-mineralogiska föreningen i Lund och Sveriges kemiska industrikontor. Programförklaringen. Enligt presentatio- nen i första numret skulle tidningen inne- hålla följande huvudpunkter (stavningen moderniserad): ”a) Kortfattade referat av det viktigare ur förhandlingarna vid Kemistsamfundet i Stockholm och de Kemiska Sektio- nerna i Uppsala och Lund. -
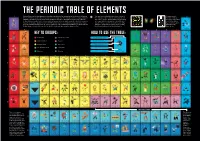
PERIODIC TABLE of ELEMENTS 1 with Well Over One Hundred Elements, Scientists Organise Them Into Groups Based on the Way They Behave
THE PERIODIC TABLE OF ELEMENTS 1 With well over one hundred elements, scientists organise them into groups based on the way they behave. You may have noticed that the colour of some of Discover more in 18 This is called the periodic table. It is just one of several ways that the elements can be arranged, and makes the elements at the end of the table don’t match The Extraordinary 1 1.008 2 4.003 navigating the building blocks of the Universe simpler. Each tile on this table represents one of the 118 any of the colours in the key. This is because they Elements © Big Picture elements that we know of so far, colour-coded into groups that have similar properties. Vertical columns in are so unstable they disappear in the blink of an Press 2020 Written the table are known as ‘groups’ (numbered 1-18) with the mass of an atom increasing as you move down a eye after being created! This makes it incredibly by Colin Stuart, with I H He group. Horizontal rows are known as ‘periods’ (labelled I - VII). Elements get heavier as you move from left difficult to study their properties and accurately illustrations by to right along each period. A new electron shell is added each time you move down a period. identify which group they should belong to. Ximo Abadia Hydrogen Helium The element that fuels Lighter than air, this makes the majority of stars. 2 13 14 15 16 17 zeppelins float. 3 6.94 4 9.012 KEY TO GROUPS: HOW TO USE THE TABLE: 5 10.81 6 12.011 7 14.007 8 15.999 9 18.998 10 20.18 Li Be B N O Ne II F The average mass of the element’s C Alkali Metals Reactive Non-metals isotopes as found in nature.