DNA Methylation-Based Classification and Identification of Renal Cell
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
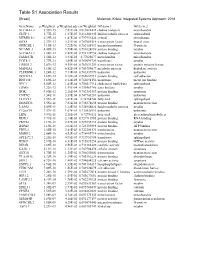
Table S1 Association Results
GeneName p.Weighted p.Weighted.adj cor.Weighted GO.term.1 GO.term.2 SLC44A1.2 3.55E-15 7.81E-08 0.819822422 choline transport mitochondria GLTP.1 5.77E-15 1.27E-07 0.816302445 lipid metabolic process sphingolipid MTMR10.1 6.39E-14 1.41E-06 0.797951424 cytosol phosphatase SOX8 2.37E-13 5.21E-06 0.787085514 transcription factor neural crest GPRC5B.1 4.19E-13 9.22E-06 0.782154937 integral membrane G-protein NCAM1.3 4.45E-13 9.79E-06 0.781624896 protein binding myelin SLC44A1.1 1.28E-12 2.82E-05 0.772132954 choline transport mitochondria FAM107B 1.56E-12 3.43E-05 0.77025677 mitochondria mitochondria UGT8.1 1.77E-12 3.89E-05 0.769099729 transferase myelin ERBB3.2 2.07E-12 4.55E-05 0.767631259 transcription factor protein tyrosine kinase MAN2A1 3.10E-12 6.82E-05 0.763759677 metabolic process hydrolase activity PLEKHH1.1 3.24E-12 7.13E-05 0.763337879 unknown unknown DOCK5.3 3.69E-12 8.12E-05 0.762087913 protein binding cell adhesion RNF130 3.69E-12 8.12E-05 0.762094156 membrane metal ion binding NPC1 6.50E-12 1.43E-04 0.756517114 cholesterol trafficking sphingolipid ERMN 7.22E-12 1.59E-04 0.755469786 actin binding myelin BOK 9.80E-12 2.16E-04 0.752383357 protein binding apoptosis CNTN2 1.54E-11 3.39E-04 0.747743281 unknown unknown ELOVL1 1.55E-11 3.41E-04 0.74764744 fatty acid sphingolipid DBNDD2 3.55E-11 7.81E-04 0.738878658 protein binding neuron projection LASS2 5.09E-11 1.12E-03 0.734954024 lipid metabolic process myelin C12orf34 7.57E-11 1.67E-03 0.730528911 unknown unknown LIPA 9.59E-11 2.11E-03 0.72786111 fatty acid glycerolipid metabolic -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Non-Coding Functional Snps Within the Arthritis-Associated TRAF1-C5 Locus
Non-Coding Functional SNPs Within the Arthritis-Associated TRAF1-C5 Locus The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Chiu, Darren Jianjhih. 2018. Non-Coding Functional SNPs Within the Arthritis-Associated TRAF1-C5 Locus. Master's thesis, Harvard Medical School. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42076544 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA ! ! Non-coding Functional SNPs within the Arthritis-associated TRAF1-C5 Locus Darren Jianjhih Chiu A Thesis Submitted to the Faculty of The Harvard Medical School in Partial Fulfillment of the Requirements for the Degree of Master of Medical Sciences in Immunology Harvard University Boston, Massachusetts. May, 2018 Thesis Advisor: Dr. Peter Nigrovic Darren Jianjhih Chiu! Non-coding functional SNPs within the arthritis-associated TRAF1-C5 locus Abstract The TRAF1-C5 locus is associated by genome-wide association studies (GWAS) with susceptibility to rheumatoid arthritis and juvenile idiopathic arthritis. Monocytes from healthy individuals with the arthritis-associated risk variant rs3761847 express lower intracellular TRAF1 protein in response to LPS and have greater LPS-induced production of IL-6 and TNF, consistent with a role in inflammatory disease. However, the functional interpretation of this finding remains challenging. Tagging SNPs identified by GWAS are often not causal themselves, but rather simply reside in close association with true functional variants. -

High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype
Supplementary Information High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype Håkon Reikvam 1,2,*, Elise Aasebø 1, Annette K. Brenner 2, Sushma Bartaula-Brevik 1, Ida Sofie Grønningsæter 2, Rakel Brendsdal Forthun 2, Randi Hovland 3,4 and Øystein Bruserud 1,2 1 Department of Clinical Science, University of Bergen, 5020, Bergen, Norway 2 Department of Medicine, Haukeland University Hospital, 5021, Bergen, Norway 3 Department of Medical Genetics, Haukeland University Hospital, 5021, Bergen, Norway 4 Institute of Biomedicine, University of Bergen, 5020, Bergen, Norway * Correspondence: [email protected]; Tel.: +55-97-50-00 J. Clin. Med. 2019, 8, x 2 of 36 Figure S1. Mutational studies in a cohort of 71 AML patients. The figure shows the number of patients with the various mutations (upper), the number of mutations in for each patient (middle) and the number of main classes with mutation(s) in each patient (lower). 2 J. Clin. Med. 2019, 8, x; doi: www.mdpi.com/journal/jcm J. Clin. Med. 2019, 8, x 3 of 36 Figure S2. The immunophenotype of primary human AML cells derived from 62 unselected patients. The expression of the eight differentiation markers CD13, CD14, CD15, CD33, CD34, CD45, CD117 and HLA-DR was investigated for 62 of the 71 patients included in our present study. We performed an unsupervised hierarchical cluster analysis and identified four patient main clusters/patient subsets. The mutational profile for each f the 62 patients is also given (middle), no individual mutation of main class of mutations showed any significant association with any of the for differentiation marker clusters (middle). -

Application of Microrna Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease
Article Application of microRNA Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease Jennifer L. Major, Rushita A. Bagchi * and Julie Pires da Silva * Department of Medicine, Division of Cardiology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; [email protected] * Correspondence: [email protected] (R.A.B.); [email protected] (J.P.d.S.) Supplementary Tables Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 www.mdpi.com/journal/mps Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 2 of 25 Table 1. List of all hsa-miRs identified by Human microRNA Disease Database (HMDD; v3.2) analysis. hsa-miRs were identified using the term “genetics” and “circulating” as input in HMDD. Targets CAD hsa-miR-1 Targets IR injury hsa-miR-423 Targets Obesity hsa-miR-499 hsa-miR-146a Circulating Obesity Genetics CAD hsa-miR-423 hsa-miR-146a Circulating CAD hsa-miR-149 hsa-miR-499 Circulating IR Injury hsa-miR-146a Circulating Obesity hsa-miR-122 Genetics Stroke Circulating CAD hsa-miR-122 Circulating Stroke hsa-miR-122 Genetics Obesity Circulating Stroke hsa-miR-26b hsa-miR-17 hsa-miR-223 Targets CAD hsa-miR-340 hsa-miR-34a hsa-miR-92a hsa-miR-126 Circulating Obesity Targets IR injury hsa-miR-21 hsa-miR-423 hsa-miR-126 hsa-miR-143 Targets Obesity hsa-miR-21 hsa-miR-223 hsa-miR-34a hsa-miR-17 Targets CAD hsa-miR-223 hsa-miR-92a hsa-miR-126 Targets IR injury hsa-miR-155 hsa-miR-21 Circulating CAD hsa-miR-126 hsa-miR-145 hsa-miR-21 Targets Obesity hsa-mir-223 hsa-mir-499 hsa-mir-574 Targets IR injury hsa-mir-21 Circulating IR injury Targets Obesity hsa-mir-21 Targets CAD hsa-mir-22 hsa-mir-133a Targets IR injury hsa-mir-155 hsa-mir-21 Circulating Stroke hsa-mir-145 hsa-mir-146b Targets Obesity hsa-mir-21 hsa-mir-29b Methods Protoc. -
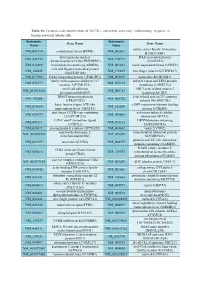
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

In Cancer Cells by Heat Shock Stimulation
583-593.qxd 19/7/2010 11:13 Ì ™ÂÏ›‰·583 INTERNATIONAL JOURNAL OF ONCOLOGY 37: 583-593, 2010 583 Induction of HITS, a newly identified family with sequence similarity 107 protein (FAM107B), in cancer cells by heat shock stimulation HIDEO NAKAJIMA1, YASUHITO ISHIGAKI2, QI-SHENG XIA1, TAKAYUKI IKEDA3, YOSHINO YOSHITAKE3, HIDETO YONEKURA3, TAKAYUKI NOJIMA4, TAKUJI TANAKA4, HISANORI UMEHARA5, NAOHISA TOMOSUGI2, TAKANOBU TAKATA2, TAKEO SHIMASAKI1, NAOKI NAKAYA1, ITARU SATO1, KAZUYUKI KAWAKAMI6, KEITA KOIZUMI7, TOSHINARI MINAMOTO6 and YOSHIHARU MOTOO1 1Department of Medical Oncology, 2Medical Research Institute and Departments of 3Biochemistry, 4Pathology and 5Hematology and Immunology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Kahoku, Ishikawa 920-0293; 6Division of Translational and Clinical Oncology, Cancer Research Institute and 7Department of Biophysical Genetics, Graduate School of Medical Science, Kanazawa University, 13-1 Takara-machi, Kanazawa, Ishikawa 920-0934, Japan Received March 31, 2010; Accepted May 14, 2010 DOI: 10.3892/ijo_00000707 Abstract. The Family with sequence similarity 107 (FAM107) Introduction possesses an N-terminal domain of unknown function (DUF1151) that is highly conserved beyond species. In human, The Family with sequence similarity 107 (FAM107) possesses FAM107A termed TU3A/DRR1 has been reported as a can- an N-terminal domain of unknown function (DUF1151) that didate tumor suppressor gene which expression is down- is conserved beyond species including mammalian, Xenopus, regulated in several types of cancer, however no studies have fish and Drosophila, and shows no homology match to investigated the other family protein, FAM107B. In the other functional conserved domains (http://www.ncbi.nlm. present study, we designated FAM107B as heat shock- nih.gov/structure/cdd/cdd.shtml). -

LADON, a Natural Antisense Transcript of NODAL, Promotes Metastasis in Melanoma by Repressing NDRG1
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.09.032375; this version posted April 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. LADON, a natural antisense transcript of NODAL, promotes metastasis in melanoma by repressing NDRG1 Dutriaux Annie1, Diazzi Serena1, Caburet Sandrine2, Bresesti Chiara1, Hardouin Sylvie1, Deshayes Frédérique3, Collignon Jérôme1,* and Flagiello Domenico1* 1: Université de Paris, CNRS, Institut Jacques Monod, Regulation of Cell-Fate Specification team, Paris 75013, France 2: Université de Paris, CNRS, Institut Jacques Monod, Molecular Oncology and Ovarian Pathologies team, Paris 75013, France 3: Université de Paris, CNRS, Institut Jacques Monod, Morphogenesis, Homeostasis and Pathologies team, Paris 75013, France *Correspondence: [email protected]; [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.09.032375; this version posted April 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary The TGFb family member NODAL, primarily known for its role during embryonic development, has also been associated with tumor progression in a number of cancers. Some of the evidence supporting its involvement in melanoma has appeared contradictory, suggesting that NODAL in this context might rely on a non-canonical mode of signaling. To investigate this possibility we studied how a deletion of NODAL affected cell behavior in a metastatic melanoma cell line. The mutation does prevent melanoma cells from acquiring an invasive behavior. -

Heparanase Overexpression Induces Glucagon Resistance and Protects
Page 1 of 85 Diabetes Heparanase overexpression induces glucagon resistance and protects animals from chemically-induced diabetes Dahai Zhang1, Fulong Wang1, Nathaniel Lal1, Amy Pei-Ling Chiu1, Andrea Wan1, Jocelyn Jia1, Denise Bierende1, Stephane Flibotte1, Sunita Sinha1, Ali Asadi2, Xiaoke Hu2, Farnaz Taghizadeh2, Thomas Pulinilkunnil3, Corey Nislow1, Israel Vlodavsky4, James D. Johnson2, Timothy J. Kieffer2, Bahira Hussein1 and Brian Rodrigues1 1Faculty of Pharmaceutical Sciences, UBC, 2405 Wesbrook Mall, Vancouver, BC, Canada V6T 1Z3; 2Department of Cellular & Physiological Sciences, Life Sciences Institute, UBC, 2350 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3; 3Department of Biochemistry and Molecular Biology, Faculty of Medicine, Dalhousie University, 100 Tucker Park Road, Saint John, NB, Canada E2L 4L5; 4Cancer and Vascular Biology Research Center, Rappaport Faculty of Medicine, Technion, Haifa, Israel 31096 Running Title: Heparanase overexpression and the pancreatic islet Corresponding author: Dr. Brian Rodrigues Faculty of Pharmaceutical Sciences University of British Columbia, 2405 Wesbrook Mall, Vancouver, B.C., Canada V6T 1Z3 TEL: (604) 822-4758; FAX: (604) 822-3035 E-mail: [email protected] Key Words: Heparanase, heparan sulfate proteoglycan, glucose homeostasis, glucagon resistance, pancreatic islet, STZ Word Count: 4761 Total Number of DiabetesFigures: Publish 6 Ahead of Print, published online October 7, 2016 Diabetes Page 2 of 85 Abstract Heparanase, a protein with enzymatic and non-enzymatic properties, contributes towards disease progression and prevention. In the current study, a fortuitous observation in transgenic mice globally overexpressing heparanase (hep-tg) was the discovery of improved glucose homeostasis. We examined the mechanisms that contribute towards this improved glucose metabolism. Heparanase overexpression was associated with enhanced GSIS and hyperglucagonemia, in addition to changes in islet composition and structure.