Amy Elizabeth Defnet Contact Information: [email protected] Degree and Date to Be Conferred: Ph.D
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . DIP2A (L-16): sc-67555 BACKGROUND APPLICATIONS DIP2A (Disco-interacting protein 2 homolog A), also known as DIP2, is a 1,571 DIP2A (L-16) is recommended for detection of DIP2A of human origin by amino acid nuclear protein. It is one of three human homologs (DIP2A, DIP2B Western Blotting (starting dilution 1:200, dilution range 1:100-1:1000), and DIP2C) of the Drosophila dip2 (disconnected-interacting protein 2) protein. immunoprecipitation [1-2 µg per 100-500 µg of total protein (1 ml of cell In Drosophila , dip2 interacts with disco, a protein required for neuronal con - lysate)], immunofluorescence (starting dilution 1:50, dilution range 1:50- nections in the visual systems of larvae and adults. The closest vertebrate 1:500) and solid phase ELISA (starting dilution 1:30, dilution range 1:30- homologs to disco are the basonuclin genes. In mice, DIP2 homologs show 1:3000). restricted expression to the brain. This suggests that, similar to the function DIP2A (L-16) is also recommended for detection of DIP2A, also designated of Drosphila dip2, vertebrate DIP2 homologs may play a role in the develop - Disco-interacting protein 2 homolog A, in additional species, including ment of the nervous system. Expressed ubiquitously with highest expression canine. in the brain, DIP2A is thought to function in signaling throughout the central nervous system by providing positional clues for axon patterning and pathfind - Suitable for use as control antibody for DIP2A siRNA (h): sc-62212, DIP2A ing . Four isoforms of DIP2A exist due to alternative splicing events. -

Diagnosing Platelet Secretion Disorders: Examples Cases
Diagnosing platelet secretion disorders: examples cases Martina Daly Department of Infection, Immunity and Cardiovascular Disease, University of Sheffield Disclosures for Martina Daly In compliance with COI policy, ISTH requires the following disclosures to the session audience: Research Support/P.I. No relevant conflicts of interest to declare Employee No relevant conflicts of interest to declare Consultant No relevant conflicts of interest to declare Major Stockholder No relevant conflicts of interest to declare Speakers Bureau No relevant conflicts of interest to declare Honoraria No relevant conflicts of interest to declare Scientific Advisory No relevant conflicts of interest to declare Board Platelet granule release Agonists (FIIa, Collagen, ADP) Signals Activation Shape change Membrane fusion Release of granule contents Platelet storage organelles lysosomes a granules Enzymes including cathepsins Adhesive proteins acid hydrolases Clotting factors and their inhibitors Fibrinolytic factors and their inhibitors Proteases and antiproteases Growth and mitogenic factors Chemokines, cytokines Anti-microbial proteins Membrane glycoproteins dense (d) granules ADP/ATP Serotonin histamine inorganic polyphosphate Platelet a-granule contents Type Prominent components Membrane glycoproteins GPIb, aIIbb3, GPVI Clotting factors VWF, FV, FXI, FII, Fibrinogen, HMWK, FXIII? Clotting inhibitors TFPI, protein S, protease nexin-2 Fibrinolysis components PAI-1, TAFI, a2-antiplasmin, plasminogen, uPA Other protease inhibitors a1-antitrypsin, a2-macroglobulin -
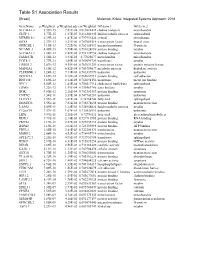
Table S1 Association Results
GeneName p.Weighted p.Weighted.adj cor.Weighted GO.term.1 GO.term.2 SLC44A1.2 3.55E-15 7.81E-08 0.819822422 choline transport mitochondria GLTP.1 5.77E-15 1.27E-07 0.816302445 lipid metabolic process sphingolipid MTMR10.1 6.39E-14 1.41E-06 0.797951424 cytosol phosphatase SOX8 2.37E-13 5.21E-06 0.787085514 transcription factor neural crest GPRC5B.1 4.19E-13 9.22E-06 0.782154937 integral membrane G-protein NCAM1.3 4.45E-13 9.79E-06 0.781624896 protein binding myelin SLC44A1.1 1.28E-12 2.82E-05 0.772132954 choline transport mitochondria FAM107B 1.56E-12 3.43E-05 0.77025677 mitochondria mitochondria UGT8.1 1.77E-12 3.89E-05 0.769099729 transferase myelin ERBB3.2 2.07E-12 4.55E-05 0.767631259 transcription factor protein tyrosine kinase MAN2A1 3.10E-12 6.82E-05 0.763759677 metabolic process hydrolase activity PLEKHH1.1 3.24E-12 7.13E-05 0.763337879 unknown unknown DOCK5.3 3.69E-12 8.12E-05 0.762087913 protein binding cell adhesion RNF130 3.69E-12 8.12E-05 0.762094156 membrane metal ion binding NPC1 6.50E-12 1.43E-04 0.756517114 cholesterol trafficking sphingolipid ERMN 7.22E-12 1.59E-04 0.755469786 actin binding myelin BOK 9.80E-12 2.16E-04 0.752383357 protein binding apoptosis CNTN2 1.54E-11 3.39E-04 0.747743281 unknown unknown ELOVL1 1.55E-11 3.41E-04 0.74764744 fatty acid sphingolipid DBNDD2 3.55E-11 7.81E-04 0.738878658 protein binding neuron projection LASS2 5.09E-11 1.12E-03 0.734954024 lipid metabolic process myelin C12orf34 7.57E-11 1.67E-03 0.730528911 unknown unknown LIPA 9.59E-11 2.11E-03 0.72786111 fatty acid glycerolipid metabolic -

Tumor-Associated Antigens Identified Early in Mouse Mammary Tumor Development Can Be Effective Vaccine Targets
Vaccine 37 (2019) 3552–3561 Contents lists available at ScienceDirect Vaccine journal homepage: www.elsevier.com/locate/vaccine Tumor-associated antigens identified early in mouse mammary tumor development can be effective vaccine targets ⇑ Sasha E. Stanton a, , Ekram Gad a, Lauren R. Corulli a, Hailing Lu a,1, Mary L. Disis a a Cancer Vaccine Institute, University of Washington, Seattle WA, 98109, USA article info abstract Article history: Breast cancer vaccines composed of antigens identified by serological analysis of cDNA expression Received 25 June 2018 libraries (SEREX) induce antigen specific immune responses in patients but have had disappointing clin- Received in revised form 5 April 2019 ical benefits. While many attempts to modify the adjuvants and vaccine method have been tried, one Accepted 9 May 2019 issue not addressed was whether the SEREX tumor-associated antigens identified from late stages of dis- Available online 21 May 2019 ease were ideal targets. We questioned in the transgenic TgMMTV-neu mouse model whether the antigen repertoire is distinct between early and late stage breast cancer and whether the antigens identified via Keywords: SEREX from transgenic mice with early or late stage tumors would elicit differential anti-tumor effects to Breast cancer prevention address this question. Vaccine antigens Th1 Three early stage antigens, Pdhx, Stk39, and Otud6B, were identified from a SEREX screen of mice prior DNA vaccines to development of palpable lesions. Formulated into a vaccine, each early antigen inhibited tumor growth Mouse mammary tumor models (p < 0.0001). The antigens identified from mice with late stage tumors (Swap70, Gsn, and Arhgef2) were unable to inhibit tumor growth when used as vaccines (for example Gsn p = 0.26). -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Clinicopathological and Molecular Genetic Analysis of Low-Grade Glioma in Adults
A CLINICOPATHOLOGICAL AND MOLECULAR GENETIC ANALYSIS OF LOW-GRADE GLIOMA IN ADULTS Presented by ANUSHREE SINGH MSc A thesis submitted in partial fulfilment of the requirements of the University of Wolverhampton for the degree of Doctor of Philosophy Brain Tumour Research Centre Research Institute in Healthcare Sciences Faculty of Science and Engineering University of Wolverhampton November 2014 i DECLARATION This work or any part thereof has not previously been presented in any form to the University or to any other body whether for the purposes of assessment, publication or for any other purpose (unless otherwise indicated). Save for any express acknowledgments, references and/or bibliographies cited in the work, I confirm that the intellectual content of the work is the result of my own efforts and of no other person. The right of Anushree Singh to be identified as author of this work is asserted in accordance with ss.77 and 78 of the Copyright, Designs and Patents Act 1988. At this date copyright is owned by the author. Signature: Anushree Date: 30th November 2014 ii ABSTRACT The aim of the study was to identify molecular markers that can determine progression of low grade glioma. This was done using various approaches such as IDH1 and IDH2 mutation analysis, MGMT methylation analysis, copy number analysis using array comparative genomic hybridisation and identification of differentially expressed miRNAs using miRNA microarray analysis. IDH1 mutation was present at a frequency of 71% in low grade glioma and was identified as an independent marker for improved OS in a multivariate analysis, which confirms the previous findings in low grade glioma studies. -

Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17P11.2 and the Syntenic Region of the Mouse
Downloaded from genome.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Article Genes in a Refined Smith-Magenis Syndrome Critical Deletion Interval on Chromosome 17p11.2 and the Syntenic Region of the Mouse Weimin Bi,1,6 Jiong Yan,1,6 Paweł Stankiewicz,1 Sung-Sup Park,1,7 Katherina Walz,1 Cornelius F. Boerkoel,1 Lorraine Potocki,1,3 Lisa G. Shaffer,1 Koen Devriendt,4 Małgorzata J.M. Nowaczyk,5 Ken Inoue,1 and James R. Lupski1,2,3,8 Departments of 1Molecular & Human Genetics, 2Pediatrics, Baylor College of Medicine, 3Texas Children’s Hospital, Houston, Texas 77030, USA; 4Centre for Human Genetics, University Hospital Gasthuisberg, Catholic University of Leuven, B-3000 Leuven, Belgium; 5Department of Pathology and Molecular Medicine, McMaster University, Hamilton, Ontario L8S 4J9, Canada Smith-Magenis syndrome (SMS) is a multiple congenital anomaly/mental retardation syndrome associated with behavioral abnormalities and sleep disturbance. Most patients have the same ∼4 Mb interstitial genomic deletion within chromosome 17p11.2. To investigate the molecular bases of the SMS phenotype, we constructed BAC/PAC contigs covering the SMS common deletion interval and its syntenic region on mouse chromosome 11. Comparative genome analysis reveals the absence of all three ∼200-kb SMS-REP low-copy repeats in the mouse and indicates that the evolution of SMS-REPs was accompanied by transposition of adjacent genes. Physical and genetic map comparisons in humans reveal reduced recombination in both sexes. Moreover, by examining the deleted regions in SMS patients with unusual-sized deletions, we refined the minimal Smith-Magenis critical region (SMCR) to an ∼1.1-Mb genomic interval that is syntenic to an ∼1.0-Mb region in the mouse. -

Mouse Vipas39 Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Vipas39 Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Vipas39 conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Vipas39 gene (NCBI Reference Sequence: NM_001142580 ; Ensembl: ENSMUSG00000021038 ) is located on Mouse chromosome 12. 20 exons are identified, with the ATG start codon in exon 2 and the TAA stop codon in exon 20 (Transcript: ENSMUST00000072744). Exon 3 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Vipas39 gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-213E8 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Mice homozygous for a conditional allele activated by an inducible cre exhibit dry and scaly skin, hair loss, and defects in tail tendon collagen I structure. Exon 3 starts from about 6.38% of the coding region. The knockout of Exon 3 will result in frameshift of the gene. The size of intron 2 for 5'-loxP site insertion: 3170 bp, and the size of intron 3 for 3'-loxP site insertion: 1410 bp. The size of effective cKO region: ~603 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 3 4 20 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Exon of mouse Vipas39 Homology arm cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Loss of DIP2C in RKO Cells Stimulates Changes in DNA
Larsson et al. BMC Cancer (2017) 17:487 DOI 10.1186/s12885-017-3472-5 RESEARCH ARTICLE Open Access Loss of DIP2C in RKO cells stimulates changes in DNA methylation and epithelial- mesenchymal transition Chatarina Larsson1, Muhammad Akhtar Ali1,2, Tatjana Pandzic1, Anders M. Lindroth3, Liqun He1,4 and Tobias Sjöblom1* Abstract Background: The disco-interacting protein 2 homolog C (DIP2C) gene is an uncharacterized gene found mutated in a subset of breast and lung cancers. To understand the role of DIP2C in tumour development we studied the gene in human cancer cells. Methods: We engineered human DIP2C knockout cells by genome editing in cancer cells. The growth properties of the engineered cells were characterised and transcriptome and methylation analyses were carried out to identify pathways deregulated by inactivation of DIP2C. Effects on cell death pathways and epithelial-mesenchymal transition traits were studied based on the results from expression profiling. Results: Knockout of DIP2C in RKO cells resulted in cell enlargement and growth retardation. Expression profiling revealed 780 genes for which the expression level was affected by the loss of DIP2C, including the tumour-suppressor encoding CDKN2A gene, the epithelial-mesenchymal transition (EMT) regulator-encoding ZEB1,andCD44 and CD24 that encode breast cancer stem cell markers. Analysis of DNA methylation showed more than 30,000 sites affected by differential methylation, the majority of which were hypomethylated following loss of DIP2C. Changes in DNA methylation at promoter regions were strongly correlated to changes in gene expression, and genes involved with EMT and cell death were enriched among the differentially regulated genes. -

Overexpressed WDR3 Induces the Activation of Hippo Pathway by Interacting with GATA4 in Pancreatic Cancer
Overexpressed WDR3 Induces the Activation of Hippo Pathway by Interacting with GATA4 in Pancreatic Cancer Wenjie Su Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Shikai Zhu Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Kai Chen Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Hongji Yang Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Mingwu Tian Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Qiang Fu Massachusetts General Hospital Ganggang Shi University of British Columbia School of Human Kinetics: The University of British Columbia School of Kinesiology Shijian Feng University of British Columbia School of Human Kinetics: The University of British Columbia School of Kinesiology Dianyun Ren Wuhan Union Hospital Xin Jin Wuhan Union Hospital Chong Yang ( [email protected] ) Sichuan Provincial People's Hospital: Sichuan Academy of Medical Sciences and Sichuan People's Hospital Page 1/34 Research Keywords: Pancreatic Cancer, WDR3, GATA4, YAP1, Hippo Signaling Pathway Posted Date: November 13th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-104564/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on March 1st, 2021. See the published version at https://doi.org/10.1186/s13046-021-01879-w. Page 2/34 Abstract Background: WD repeat domain 3 (WDR3) is involved in a variety of cellular processes including gene regulation, cell cycle progression, signal transduction and apoptosis. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US).