Identification, by Systematic RNA Sequencing, of Novel Candidate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SRC Antibody - N-Terminal Region (ARP32476 P050) Data Sheet
SRC antibody - N-terminal region (ARP32476_P050) Data Sheet Product Number ARP32476_P050 Product Name SRC antibody - N-terminal region (ARP32476_P050) Size 50ug Gene Symbol SRC Alias Symbols ASV; SRC1; c-SRC; p60-Src Nucleotide Accession# NM_005417 Protein Size (# AA) 536 amino acids Molecular Weight 60kDa Product Format Lyophilized powder NCBI Gene Id 6714 Host Rabbit Clonality Polyclonal Official Gene Full Name V-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (avian) Gene Family SH2D This is a rabbit polyclonal antibody against SRC. It was validated on Western Blot by Aviva Systems Biology. At Aviva Systems Biology we manufacture rabbit polyclonal antibodies on a large scale (200-1000 Description products/month) of high throughput manner. Our antibodies are peptide based and protein family oriented. We usually provide antibodies covering each member of a whole protein family of your interest. We also use our best efforts to provide you antibodies recognize various epitopes of a target protein. For availability of antibody needed for your experiment, please inquire (). Peptide Sequence Synthetic peptide located within the following region: QTPSKPASADGHRGPSAAFAPAAAEPKLFGGFNSSDTVTSPQRAGPLAGG This gene is highly similar to the v-src gene of Rous sarcoma virus. This proto-oncogene may play a role in the Description of Target regulation of embryonic development and cell growth. SRC protein is a tyrosine-protein kinase whose activity can be inhibited by phosphorylation by c-SRC kinase. Mutations in this gene could be involved in the -

SMARCB1/INI1 Genetic Inactivation Is Responsible for Tumorigenic Properties of Epithelioid Sarcoma Cell Line VAESBJ
Published OnlineFirst April 10, 2013; DOI: 10.1158/1535-7163.MCT-13-0005 Molecular Cancer Cancer Therapeutics Insights Therapeutics SMARCB1/INI1 Genetic Inactivation Is Responsible for Tumorigenic Properties of Epithelioid Sarcoma Cell Line VAESBJ Monica Brenca1, Sabrina Rossi3, Erica Lorenzetto1, Elena Piccinin1, Sara Piccinin1, Francesca Maria Rossi2, Alberto Giuliano1, Angelo Paolo Dei Tos3, Roberta Maestro1, and Piergiorgio Modena1 Abstract Epithelioid sarcoma is a rare soft tissue neoplasm that usually arises in the distal extremities of young adults. Epithelioid sarcoma presents a high rate of recurrences and metastases and frequently poses diagnostic dilemmas. We previously reported loss of tumor suppressor SMARCB1 protein expression and SMARCB1 gene deletion in the majority of epithelioid sarcoma cases. Unfortunately, no appropriate preclinical models of such genetic alteration in epithelioid sarcoma are available. In the present report, we identified lack of SMARCB1 protein due to a homozygous deletion of exon 1 and upstream regulatory region in epithelioid sarcoma cell line VAESBJ. Restoration of SMARCB1 expression significantly affected VAESBJ cell proliferation, anchorage-independent growth, and cell migration properties, thus supporting the causative role of SMARCB1 loss in epithelioid sarcoma pathogenesis. We investigated the translational relevance of this genetic back- ground in epithelioid sarcoma and showed that SMARCB1 ectopic expression significantly augmented VAESBJ sensitivity to gamma irradiation and acted synergistically with flavopiridol treatment. In VAESBJ, both activated ERBB1/EGFR and HGFR/MET impinged on AKT and ERK phosphorylation. We showed a synergistic effect of combined inhibition of these 2 receptor tyrosine kinases using selective small-molecule inhibitors on cell proliferation. These observations provide definitive support to the role of SMARCB1 inactivation in the pathogenesis of epithelioid sarcoma and disclose novel clues to therapeutic approaches tailored to SMARCB1-negative epithelioid sarcoma. -

The PTEN Tumor Suppressor Gene in Soft Tissue Sarcoma
cancers Review The PTEN Tumor Suppressor Gene in Soft Tissue Sarcoma Sioletic Stefano 1,* and Scambia Giovanni 2,3 1 UOC Anatomia Patologica, San Camillo De Lellis, 02100 Rieti, Italy 2 UOC di Ginecologia Oncologica, Dipartimento di Scienze della Salute della Donna e del Bambino e di Sanità Pubblica, Fondazione Policlinico Agostino Gemelli IRCCS, Largo A. Gemelli 8, 00168 Rome, Italy 3 Istituto di Clinica Ostetrica e Ginecologica, Università Cattolica del Sacro Cuore, Largo F. Vito 1, 00168 Rome, Italy * Correspondence: [email protected] Received: 15 June 2019; Accepted: 8 August 2019; Published: 14 August 2019 Abstract: Soft tissue sarcoma (STS) is a rare malignancy of mesenchymal origin classified into more than 50 different subtypes with distinct clinical and pathologic features. Despite the poor prognosis in the majority of patients, only modest improvements in treatment strategies have been achieved, largely due to the rarity and heterogeneity of these tumors. Therefore, the discovery of new prognostic and predictive biomarkers, together with new therapeutic targets, is of enormous interest. Phosphatase and tensin homolog (PTEN) is a well-known tumor suppressor that commonly loses its function via mutation, deletion, transcriptional silencing, or protein instability, and is frequently downregulated in distinct sarcoma subtypes. The loss of PTEN function has consequent alterations in important pathways implicated in cell proliferation, survival, migration, and genomic stability. PTEN can also interact with other tumor suppressors and oncogenic signaling pathways that have important implications for the pathogenesis in certain STSs. The aim of the present review is to summarize the biological significance of PTEN in STS and its potential role in the development of new therapeutic strategies. -
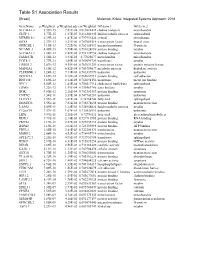
Table S1 Association Results
GeneName p.Weighted p.Weighted.adj cor.Weighted GO.term.1 GO.term.2 SLC44A1.2 3.55E-15 7.81E-08 0.819822422 choline transport mitochondria GLTP.1 5.77E-15 1.27E-07 0.816302445 lipid metabolic process sphingolipid MTMR10.1 6.39E-14 1.41E-06 0.797951424 cytosol phosphatase SOX8 2.37E-13 5.21E-06 0.787085514 transcription factor neural crest GPRC5B.1 4.19E-13 9.22E-06 0.782154937 integral membrane G-protein NCAM1.3 4.45E-13 9.79E-06 0.781624896 protein binding myelin SLC44A1.1 1.28E-12 2.82E-05 0.772132954 choline transport mitochondria FAM107B 1.56E-12 3.43E-05 0.77025677 mitochondria mitochondria UGT8.1 1.77E-12 3.89E-05 0.769099729 transferase myelin ERBB3.2 2.07E-12 4.55E-05 0.767631259 transcription factor protein tyrosine kinase MAN2A1 3.10E-12 6.82E-05 0.763759677 metabolic process hydrolase activity PLEKHH1.1 3.24E-12 7.13E-05 0.763337879 unknown unknown DOCK5.3 3.69E-12 8.12E-05 0.762087913 protein binding cell adhesion RNF130 3.69E-12 8.12E-05 0.762094156 membrane metal ion binding NPC1 6.50E-12 1.43E-04 0.756517114 cholesterol trafficking sphingolipid ERMN 7.22E-12 1.59E-04 0.755469786 actin binding myelin BOK 9.80E-12 2.16E-04 0.752383357 protein binding apoptosis CNTN2 1.54E-11 3.39E-04 0.747743281 unknown unknown ELOVL1 1.55E-11 3.41E-04 0.74764744 fatty acid sphingolipid DBNDD2 3.55E-11 7.81E-04 0.738878658 protein binding neuron projection LASS2 5.09E-11 1.12E-03 0.734954024 lipid metabolic process myelin C12orf34 7.57E-11 1.67E-03 0.730528911 unknown unknown LIPA 9.59E-11 2.11E-03 0.72786111 fatty acid glycerolipid metabolic -

National Cancer Grid Management of Bone and Soft Tissue Tumors
NCG BST GUIDELINES National Cancer Grid Management of Bone and Soft Tissue Tumors 1 | P a g e Version 1, August2020 NCG BST GUIDELINES Index S.No TOPIC Page Number 1 Evaluation of suspected bone sarcoma 3 2 Evaluation of suspected soft tissue sarcoma 4 3 Evaluation of suspected metastatic bone 5 disease 4 Osteosarcoma 6 5 Ewing’s Sarcoma 9 6 Chondrosarcoma 12 7 Extremity Soft Tissue Sarcoma 14 8 Surveillance in Sarcomas 17 9 Appendix 1: Principles of Management 18 10 Appendix 2: References 25 11 Appendix 3: Imaging 31 12 Appendix 4: Biopsy for Surgeons 35 13 Appendix 5: Biopsy for Pathologists 36 14 Appendix 6: Chemotherapy for bone and soft 41 tissue sarcomas 15 Appendix 7: Radiation for bone and soft tissue 47 sarcomas Note: The guidelines have two components, Essential and optional. All work-up unless specified otherwise is Essential. Optional where applicable has been specified. 2 | P a g e Version 1, August2020 NCG BST GUIDELINES EVALUATION OF SUSPECTED BONE SARCOMA 3 | P a g e Version 1, August2020 NCG BST GUIDELINES EVALUATION OF SUSPECTED SOFT TISSUE SARCOMA 4 | P a g e Version 1, August2020 NCG BST GUIDELINES EVALUATION OF SUSPECTED METASTATIC BONE DISEASE 5 | P a g e Version 1, August2020 NCG BST GUIDELINES OSTEOSARCOMA Symptoms – swelling & pain Detailed clinical history Clinical diagnosis Workup for diagnosis Basic imaging (local & chest x-ray) & routine blood investigations (Essential) Local 3D imaging - MRI (with contrast) of entire bone with adjoining joints (Essential) OR - Local imaging - X ray and Dynamic Contrast MRI -

Edinburgh Research Explorer
Edinburgh Research Explorer International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list Citation for published version: Davenport, AP, Alexander, SPH, Sharman, JL, Pawson, AJ, Benson, HE, Monaghan, AE, Liew, WC, Mpamhanga, CP, Bonner, TI, Neubig, RR, Pin, JP, Spedding, M & Harmar, AJ 2013, 'International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands', Pharmacological reviews, vol. 65, no. 3, pp. 967-86. https://doi.org/10.1124/pr.112.007179 Digital Object Identifier (DOI): 10.1124/pr.112.007179 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: Pharmacological reviews Publisher Rights Statement: U.S. Government work not protected by U.S. copyright General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 02. Oct. 2021 1521-0081/65/3/967–986$25.00 http://dx.doi.org/10.1124/pr.112.007179 PHARMACOLOGICAL REVIEWS Pharmacol Rev 65:967–986, July 2013 U.S. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Gene Symbol Category ACAN ECM ADAM10 ECM Remodeling-Related ADAM11 ECM Remodeling-Related ADAM12 ECM Remodeling-Related ADAM15 E
Supplementary Material (ESI) for Integrative Biology This journal is (c) The Royal Society of Chemistry 2010 Gene symbol Category ACAN ECM ADAM10 ECM remodeling-related ADAM11 ECM remodeling-related ADAM12 ECM remodeling-related ADAM15 ECM remodeling-related ADAM17 ECM remodeling-related ADAM18 ECM remodeling-related ADAM19 ECM remodeling-related ADAM2 ECM remodeling-related ADAM20 ECM remodeling-related ADAM21 ECM remodeling-related ADAM22 ECM remodeling-related ADAM23 ECM remodeling-related ADAM28 ECM remodeling-related ADAM29 ECM remodeling-related ADAM3 ECM remodeling-related ADAM30 ECM remodeling-related ADAM5 ECM remodeling-related ADAM7 ECM remodeling-related ADAM8 ECM remodeling-related ADAM9 ECM remodeling-related ADAMTS1 ECM remodeling-related ADAMTS10 ECM remodeling-related ADAMTS12 ECM remodeling-related ADAMTS13 ECM remodeling-related ADAMTS14 ECM remodeling-related ADAMTS15 ECM remodeling-related ADAMTS16 ECM remodeling-related ADAMTS17 ECM remodeling-related ADAMTS18 ECM remodeling-related ADAMTS19 ECM remodeling-related ADAMTS2 ECM remodeling-related ADAMTS20 ECM remodeling-related ADAMTS3 ECM remodeling-related ADAMTS4 ECM remodeling-related ADAMTS5 ECM remodeling-related ADAMTS6 ECM remodeling-related ADAMTS7 ECM remodeling-related ADAMTS8 ECM remodeling-related ADAMTS9 ECM remodeling-related ADAMTSL1 ECM remodeling-related ADAMTSL2 ECM remodeling-related ADAMTSL3 ECM remodeling-related ADAMTSL4 ECM remodeling-related ADAMTSL5 ECM remodeling-related AGRIN ECM ALCAM Cell-cell adhesion ANGPT1 Soluble factors and receptors -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Molecular and Genetic Medicine
Bertazzi et al., J Mol Genet Med 2015, 8:2 Molecular and Genetic Medicine http://dx.doi.org/10.4172/1747-0862.1000116 Review Article Open Access Myotubularin MTM1 Involved in Centronuclear Myopathy and its Roles in Human and Yeast Cells Dimitri L. Bertazzi#, Johan-Owen De Craene# and Sylvie Friant* Department of Molecular and Cellular Genetics, UMR7156, Université de Strasbourg and CNRS, France #Authors contributed equally to this work. *Corresponding author: Friant S, Department of Molecular and Cellular Genetics, UMR7156, Université de Strasbourg and CNRS, 67084 Strasbourg, France, E-mail: [email protected] Received date: April 17, 2014; Accepted date: July 21, 2014; Published date: July 28, 2014 Copyright: © 2014 Bertazzi DL, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Mutations in the MTM1 gene, encoding the phosphoinositide phosphatase myotubularin, are responsible for the X-linked centronuclear myopathy (XLCNM) or X-linked myotubular myopathy (XLMTM). The MTM1 gene was first identified in 1996 and its function as a PtdIns3P and PtdIns(,5)P2 phosphatase was discovered in 2000. In recent years, very important progress has been made to set up good models to study MTM1 and the XLCNM disease such as knockout or knockin mice, the Labrador Retriever dog, the zebrafish and the yeast Saccharomyces cerevisiae. These helped to better understand the cellular function of MTM1 and of its four conserved domains: PH-GRAM (Pleckstrin Homology-Glucosyltransferase, Rab-like GTPase Activator and Myotubularin), RID (Rac1-Induced recruitment Domain), PTP/DSP (Protein Tyrosine Phosphatase/Dual-Specificity Phosphatase) and SID (SET-protein Interaction Domain). -

Stimulation of Endogenous GH and Interleukin-6 Receptors Selectively Activates Different Jaks and Stats, with a Stat5 Specific Synergistic Effect of Dexamethasone
301 Stimulation of endogenous GH and interleukin-6 receptors selectively activates different Jaks and Stats, with a Stat5 specific synergistic effect of dexamethasone S von Laue1, J Finidori3, M Maamra1, X-Y Shen1, S Justice1, P R M Dobson2 and R J M Ross1 1Division of Clinical Sciences, University of Sheffield, UK 2Institute for Cancer Studies, University of Sheffield, UK 3INSERM, Unité 344, Faculté de Médecine Necker, Paris, France (Requests for offprints should be addressed to S von Laue, INSERM U344, Laboratoire d’Endocrinologie Moléculaire, Faculté de Médecine Necker-Enfants Malades, 156, rue de Vaugirard, 75 730 Paris cedex 15, France; Email: [email protected]) Abstract The interaction of GH, interleukin (IL)-6 and glucocorti- Stat3 activation. In contrast, the reporter gene containing coids is likely to be important in regulating the GH- the Stat3 responsive element (SIE) was IL-6 specific. The insulin-like growth factor (IGF)-I axis. The signalling levels of gene induction by GH and IL-6 were not altered cascades activated by GH and IL-6 appear to be very by the co-stimulation with GH and IL-6, suggesting that similar, as demonstrated by studies using overexpression of there is little cross-talk at the Jak–Stat activation level the receptor and other components of the Jak-Stat and between the two cytokines. Neither GH nor IL-6 acti- mitogen-activated protein (MAP) kinase pathways. Here vated the MAP-kinase responsive serum response element we show that the human embryonic kidney cell line 293 (SRE), unless GH receptors or gp130 were overexpressed. (HEK293) expresses GH and IL-6 receptors endogenously.