Table of Contents I
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Der Frankfurter Norden: Mehr Als Nur Stadtrand
Arbeitskreis Nord der Frankfurter SPD Der Frankfurter Norden: Mehr als nur Stadtrand. Heimat für Frankfurterinnen und Frankfurter 14. November 2015 Positionspapier zur Weiterentwicklung der nördlichen Stadtteile zur Kommunalwahl 2016 Fortschreibung der Position der nördlichen Ortsvereine (Erstfassung 1993) Beschluss der gemeinsamen Mitgliederversammlung der nördlichen Ortsvereine vom 14.11.2015 1.Vorwort Frankfurt und sein Magistrat setzen sich endlich mit der Problematik des fehlenden und bezahlbaren Wohnraums auseinander. Nachdem die Stadtregierung dieses Problem über Jahre verschlafen und vernachlässigt hat, hat die SPD dieses offensichtliche Problem auf die Tagesordnung gesetzt. In Frankfurt fehlen ca. 25.000 Wohnungen – Tendenz steigend. Dies ist mitverursacht durch Schwarz- Grün z.B. wegen der Reduzierung der Zahl der Wohnungen am Riedberg und der Verhinderung von innerstädtischem Wohnungsbau am Osthafen. Dieser Druck auf den Wohnungsmarkt führt zu höheren Mieten und in seiner Folge zu Gentrifizierung. Die Versorgung der Bevölkerung mit Wohnraum ist derzeit das drängendste Problem für das Rhein- Main-Gebiet und für eine Metropole wie Frankfurt. Der nationale und internationale Zuzug hält weiterhin an. Allerdings ist nicht nur die Anzahl der fehlenden Wohnungen zu betrachten, sondern auch die Bezahlbarkeit der Wohnungen für alle Menschen und ihre Familien, die in dieser Stadt einen Arbeitsplatz haben. Dieses Problem ist den Sozialdemokratinnen und Sozialdemokraten auch im Frankfurter Norden sehr bewusst. Der überraschende Vorstoß -

Und Bodenschutzalarmplan
Gewässer- und Bodenschutzalarmplan für die Stadt Frankfurt am Main Stand 10/17 Herausgeber: Magistrat der Stadt Frankfurt am Main Umweltamt Untere Wasser- und Bodenschutzbehörde Galvanistraße 28 60486 Frankfurt am Main 1 Inhaltsverzeichnis Seite 1. Allgemeines 1.1 Gültigkeitsbereich mit Übersichtsplan 3 1.2 Alarmschema 4 1.3 Meldebogen 5 2. Zweck des Gewässerschutzalarmplanes 2.1 Geltungsbereich 7 2.2 Umweltgefährdende/wassergefährdende Stoffe 7 2.3 Meldepflicht und Meldung 7 2.4 Zuständigkeiten 8 3. Gebiete von besonderer wasserwirtschaftlicher Bedeutung 3.1 Trinkwasserschutzgebiete 10 3.1.1 Auflistung der festgesetzten und beantragten Trinkwasserschutzgebiete 10 3.1.2 Kartenausschnitte der festgesetzten Trinkwasserschutzgebiete 11 3.2 Heilquellenschutzgebiete (Auflistung) 14 3.3 Festgesetzte Überschwemmungsgebiete (Auflistung) 15 3.4 Gebiete mit Trennkanalisation (Auflistung) 16 4. Verzeichnis der Meldestellen 4.1 Meldestellen in besonderen Fällen 17 4.2 Weitere in Anspruch zu nehmende Stellen 26 Stand 05/13 2 1. Allgemeines 1.1 Gültigkeitsbereich mit Übersichtsplan 1.1.1 Der Gewässerschutz-und Bodenschutzalarmplan gilt für das Stadtgebiet Frankfurt am Main 1.1.2 Stadtgebietsgrenze Main: Linkes Ufer: Main-Kilometer 22,445 - 38,385 Rechtes Ufer: Main-Kilometer 19,685 - 46,445 1.1.3 Übersichtskarte mit angrenzenden Landkreisen und Stadtgebiet Offenbach geänderter Auszug aus: Hessen 1 : 200 000 Verwaltungsgrenzenausgabe mit Gemarkung. Hrsg.: Hessisches Landesvermessungsamt 1992 Zuständige Wasser- und Bodenschutzbehörde für die jeweiligen -

Amtsblatt 1 / 2
4811 AMTSBLATT 1 / 2 Amtsblatt für Frankfurt am Main 07. Januar 2020 · Nr. 1/2 · 151. Jahrgang Neujahrsgrußwort des Oberbürgermeisters Peter Feldmann Liebe Frankfurterinnen und Frankfurter, geschnürt, um den Klimawandel einzudämmen. Und zusammen mit den engagierten Bürgerin- Sie merken und sehen es jeden Tag, wenn sie nen und Bürgern des Radentscheids wurde das aufmerksam durch Frankfurt laufen: Diese Stadt größte Investitionsprogramm in den Fahrradver- verändert sich. Es wird gebaut, neue Straßen kehr in Frankfurt auf den Weg gebracht. entstehen, ganze Viertel. Jahr für Jahr ziehen neugierige und spannende Menschen in unse- Wir arbeiten außerdem daran, die Stadt nicht nur re Stadt. Unsere Stadt - sie soll eine Stadt für lebenswerter zu machen - sondern alle daran alle Menschen bleiben. Niemand darf sich aus- teilhaben zu lassen. Mit kostenlosen Kindergär- geschlossen fühlen, niemand alleingelassen. Ich ten, seit Februar mit einem kostenlosen Eintritt finde, das funktioniert in Frankfurt schon ziem- in Schwimmbäder für Kinder und Jugendliche, lich gut. Warum das so ist, das zeigt ein Blick mit günstigeren Fahrpreisen, mit einem Senio- zurück. renticket und einem Mietenstopp bei den städ- tischen Wohnungsbaugesellschaften soll Frank- Schon immer in ihrer Geschichte hat sich un- furt nicht nur lebenswerter werden, sondern vor sere Stadt neu erfunden - weil sie neue Ideen allem bezahlbar. Im kommenden Jahr gibt es mit zugelassen hat und Bürger rasch aufgenommen dem Kultur- und Freizeitticket und kostenfreiem und integriert hat. Gerade durch ihre Rolle als Eintritt für Kinder und Jugendliche in den Zoo, Handelsmetropole konnte Frankfurt jene Stadt Senckenberg und alle städtischen und die meis- werden, aus der schlussendlich die erste deut- ten privaten Museen einen weiteren Baustein. -

Frankfurt Am Main Kalbach-Riedberg
Frankfurt am Main Kalbach-Riedberg Informationsbroschüre 2. Auflage 2018 10-5190 SUNFL Anz 205x195li V2_Merkur_Layout 1 24.03.17 10:38 Seite 1 Wir sind mehr als ein Garten-Center • Frischemarkt • Gartenplanung • Floristik/Dekoartikel • Gartenmöbel • Saunen und Whirlpools • Gartentechnik • Kulturelle Veranstaltungen • Grills/Grillschule • Exklusive Gartenbekleidung • Café/Restaurant und natürlich Pflanzen in großer Vielfalt und Qualität Wir freuen uns auf Ihren Besuch SUNFLOWERGARTENCENTER An der A 661 · Am Martinszehnten 15 · 60437 Frankfurt Telefon 069 - 50 00 49 - 0 · www.sunflower-gartencenter.de 10-5190 SUNFL Anz 205x195li V2_Merkur_Layout 1 24.03.17 10:38 Seite 1 Wir sind mehr als ein Garten-Center Grußwort der Ortsvorsteherin • Frischemarkt • Gartenplanung • Floristik/Dekoartikel • Gartenmöbel • Saunen und Whirlpools • Gartentechnik • Kulturelle Veranstaltungen • Grills/Grillschule • Exklusive Gartenbekleidung • Café/Restaurant Liebe Mitbürgerinnen und Mitbürger, wir haben unsere Informationsbroschüre aktualisiert. Das kommt und natürlich Pflanzen in großer Vielfalt und Qualität nicht von ungefähr, denn in den vergangenen Jahren ist viel in unserem Ortsbezirk passiert. Kalbach und Riedberg sind ge- wachsen und die Infrastruktur ebenfalls. Es war ein langer und spannender Weg. Sowohl für das alte, hier wohnen, wertvolle Hinweise und interessante Informatio- ehemals dörfliche Kalbach, dessen Ursprung im 8. Jahrhundert nen in dieser Broschüre finden. Neben der Darstellung der Kal- liegt, wie für den Riedberg als Frankfurts größte und jüngste bacher und Riedberger Geschichte finden Sie mit den Auflistun- Stadterweiterung. Ein neuer, moderner und urbaner Stadtteil, gen über die Verwaltung, Kirchen, Schulen, Kitas und Vereine, der an den naturwissenschaftlichen Campus der Goethe-Uni- gemeinsam mit den Hinweisen zum Geschäftsleben, ein aktuel- versität angrenzt, aber doch von attraktiven Grünflächen durch- les und lebendiges Abbild unseres Stadtteils. -

Gerichtsvollzieher/Innen Bei Dem Amtsgericht Frankfurt Am Main
Schiedsämter in dem Bezirk des Amtsgericht Frankfurt am Main Stand: 8. Februar 2021 a) Stadtbezirk Stadtverwaltung Frankfurt am Main Amt 09 60275 Frankfurt am Main Schiedsamt und Schiedsperson Telefon und Adresse Zuständigkeit Sprechzeiten Frankfurt 1 Haus Gallus Schiedsperson - Gallus Frankenallee 111 Norbert Mo. 18.00 – 19.30 h - Gutleutviertel Clubraum 2 Hetterich 069/734657 - Innenstadt Frankfurt 2 Bockenheimer Treff Schiedsperson Nach telefonischer - Bockenheim Schwälmer Straße Dietmar Vereinbarung - Westend 28 Junghans 069/751382 Frankfurt 3 Schiedsperson Nach telefonischer - Nordend Richard Gerth Vereinbarung 069/435178 Frankfurt 4 Schiedsperson Nach telefonischer - Bornheim Richard Gerth Vereinbarung - Ostend 069/435178 Frankfurt 5a Haus Südbahnhof, Schiedsperson Di. 18.00 – 19.00 h - Oberrad Hedderichstraße 51 Regina Rumohr 069/627218 - Sachsenhausen Frankfurt 5b Heimatmuseum Schiedsperson Do. 18.00 – 19.00 h - Niederrad Niederrad, Dieter Günther 069/6787865 Schwanheimer Straße 17 Frankfurt 6a Autogenstraße 19 Schiedsperson Mo. 18.00 – 19.00 h - Goldstein Stefan Brand 0178/5115580 - Griesheim - Nied - Schwanheim Frankfurt 6b Schiedsperson Nach telefonischer - Sindlingen Michael Vereinbarung - Zeilsheim Streubel 0171/6806210 Frankfurt 6c Bolongaropalast Schiedsperson Mo. 17.00 – 18.00 - Höchst Zimmer 123 Hans-Günter 069/312495 - Sossenheim Bolongarostraße 109 Neidel 0170/5308522 - Unterliederbach Frankfurt 7 Auguste-Oberwinter- Schiedsperson Mo. 18.00 – 19.00 h - Hausen Haus Thomas Fischer 069/785622 - Praunheim Burgfriedenstraße -

The Beginnings of Capitalism in Central Europe of Central Europe from 1600 to 1700 Is Separated from the Development in All of Europe and from Western Europe
PART II Cyril Levitt - 9781433172090 Downloaded from PubFactory at 09/25/2021 04:58:54PM via free access Cyril Levitt - 9781433172090 Downloaded from PubFactory at 09/25/2021 04:58:54PM via free access CHAPTER THREE Labour Processes in Central Europe, 15th–17th Centuries 3.1 The Population and Its Numbers The population of Central Europe from the 15th to the 17th century was for the most part based in the countryside, and its main occupation was tied up with agriculture. The total population of Central Europe in 1500 amounted to around 12 million, of which the country population was 9 million or about 75% of the whole. The town population counted 3 million or about 25% of the total. The country population includes the peasants, workers on the land as well as the administration, people of the cloth, servants, traders, the military. Customarily one reckoned that those who were not peasants constituted 5 to 10% of the total pop- ulation of the countryside. The town population included embossers, iron workers, and miners; yet their labour was not plied everywhere in cities or small towns. The numbers offer a rough idea; the division of the totality in the city and in the coun- try is likewise imprecise, for some iron works were then country based. In 1300, the entire population of Western Europe, as we define it inTable 1 , counted 43 million and in 1500 roughly the same number. No increase in pop- ulation was attributed to this region between 1300 and 1500; the stagnation was explained by the effects of the plague, especially of the Black Death around 1347/52, and by war.1 In 1600, the total population of Central Europe rose to 15 Million and in parts of Western Europe to 54 Million. -

Report Frankfurt Am Main - the Metropolis on the River Main with an International Format, a Global City at the Centre of Europe
RESIDENTIAL MARKET FRANKFURTREPORT FRANKFURT AM MAIN - THE METROPOLIS ON THE RIVER MAIN WITH AN INTERNATIONAL FORMAT, A GLOBAL CITY AT THE CENTRE OF EUROPE FRANKFURT AM MAIN A CITY WHICH IS GROWING HORIZONTALLY AND VERTICALLY, INTEGRATING MEETING THE DEMAND FOR LIVING SPACE WITH MODERN CONCEPTS! RESIDENTIAL MARKETREPORTFRANKFURT 3 BOOMTOWN 5,722,000 759,000 2,300,000 Population of the Rhine- Population of Population of Frankfurt FRANKFURT am Main Main metropolitan region Frankfurt am Main am Main and its suburbs The most international city in Germany, the largest financial centre in continental Europe & the fastest growing major city 2019: 759,000 +13.3% +7.1% P O P U L +4.4% A 2035: +2.4% TOP 4 T POPULATION GROWTH IO N FORECAST 2019–2035 (%) GR OWTH 860,000 HAMBURG BERLIN MUNICH FRANKFURT AM MAIN POPULATION GROWTH IN THE FRANKFURT RHINE/MAIN METROPOLITAN REGION FORECAST 6,100 Frankfurt am main IS THE CITY WITH BY FAR THE 5,900 STRONGEST POPULATION GROWTH FORECAST AMONG 5,700 THE TOP 4 IN GERMANY. IN ADDITION, THE RHINE-MAIN thousandsin REGION AS A WHOLE IS ALSO GROWING AT AN ABOVE- 5,500 AVERAGE RATE. 5,300 1999 2003 2007 2011 2015 2019 2023 2027 2031 2035 Source: Oxford Economics, Stadt Frankfurt am Main 4 5 FRANKFURT NO.1 Financial Centre AM MAIN Continental Europe GLOBAL FINANCIAL CENTRE 311 destinations in 97 countries BREXIT-WINNER 70.5 M Approx. 25 applications to Passengers in 2019 BaFin for new banking licences >1,400 NORDRHEIN-WESTFALEN FLIGHTS departures and arrivals daily HESSEN INTERNATIONAL REACH 1 of 30 global gateway cities 5 MOTORWAYS & 6 MAJOR TRUNK ROADS link Frankfurt with SECTOR MIX TALENT BASE the whole of Europe Frankfurt am Main is regarded as the job 27 Universities and other motor of the Rhine-Main area and has by institutions of higher education, far the highest office employment rate in Frankfurt am Main 50 educational facilities with Wiesbaden Germany at over 50%. -
Informationen, Beratung Und Hilfe in Frankfurt Am Main
Herausgeber: INFORMATIONEN, BERATUNG UND HILFE IN FRANKFURT AM MAIN Alkohol Cannabis Kokain Synthetische Drogen Medikamente Glücksspiel INFORMATIONEN, BERATUNG UND HILFE IN FRANKFURT AM MAIN Internet & Medien Informationen, Beratung und Hilfe Die aufgeführten Beratungsstellen bieten Ihnen als Betroffene, Angehörige oder als Fachkraft folgende Hilfen an: · Informationen zu psychoaktiven Substanzen und Verhaltenssüchten · Beratung · Vermittlung in weiterführende Hilfsangebote Die Beraterinnen und Berater unterstützen Sie und begleiten Sie auf Ihrem Weg. Die Beraterinnen und Berater unterliegen der Schweigepflicht. Die Gespräche sind kostenfrei. 3 Alkohol Cannabis Kokain Drogen Synthetische Medikamente Glücksspiel & Medien Internet Übersicht und Angebote 7 Jugend- und Drogenberatung Höchst 8 Fachstelle Nord / drop in 9 Jugendberatung und Suchthilfe Am Merianplatz 10 Jugendberatung und Suchthilfe Sachsenhausen / Haus der Beratung 12 Evangelische Suchtberatung 13 Suchtberatung im Evangelischen Zentrum für Beratung in Höchst 14 Caritas – Fachambulanz für Suchtkranke 15 CALLA – Beratungsbüro für suchtmittelabhängige Frauen 16 Suchtberatungsstelle Seckbach 17 Fachstelle Sucht – Prävention und Therapie 18 Beratungsstelle Sucht im Alter 19 Telefonische Beratung und Drogennotruf 20 Online-Beratung 21 Selbsthilfe-Kontaktstelle Frankfurt 4 5 Beratung in Frankfurt Zuständigkeit der Jugend- und Drogenberatungsstellen Auf den folgenden Seiten finden Sie im Überblick alle Angebote und Kontaktadressen der Jugend-, Drogen- und Suchtberatungsstellen in Westen: -
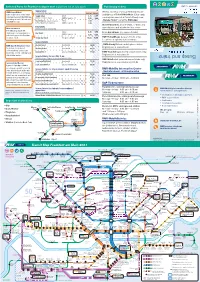
Buses and Trains In
Selected Fares for Frankfurt & airport 2021 (valid from 1st of July 2021) Purchasing tickets 1.7.2021 from valid and Tickets and and Tickets and s Transit Maps Maps Transit Transit Single ticket Weekly, monthly or annual RMV tickets are Maps Transit RMV-PrepaidRabatt For immediate travel only Frankfurt & Airport Save 20 percent on every single available as eTicket RheinMain (Chip cards Single ticket Adults 2,75 5,10 ticket purchase with the RMV-App Valid for 1 trip incl. change Children aged 6 - 14 1,55 3,00 can be purchased at all ticket offices) or as by loading at least € 40 onto your Adults „Handy-Ticket“ using the RMV-App meinRMV account. Short-trip Max. 2km, destinations 1,50 listed at stops/ticket machines Children aged 6 - 14 1,00 Ticket machines at all S-Bahn, U-Bahn and Day tickets tram stations and at selected bus stops Ideal for families! Valid until 5 am on following day The RMV group day ticket Adults 5,35 9,95 entitles up to five passengers to Day ticket From bus drivers (no season tickets) Children aged 6 - 14 unlimited travel within Frankfurt 3,00 5,85 for only € 11,50. Group day ticket For up to 5 people 11,50 16,95 VGF-TicketShops (season tickets only) Locations at vgf-ffm.de/ticketshops Season tickets „RMV-HandyTicket“ mobile phone ticket RMV-App: Mobile phone ticket Weekly ticket transferable 26,80 Registration at www.rmv.de 2021 2021 2021 No change, no queues at Monthly ticket transferable 93,10 2021 in one in in one in ticket-machines, and no fare- Annual ticket personal or transferable 12 × 77,60 RMV-TicketShop (selected season tickets only) knowledge needed. -

Bildungsraum Grüngürtel
Projektskizze Bildungsraum GrünGürtel UMWELTLERNEN IN FRANKFURT AM MAIN E.V. im Auftrag Dezernat für Bildung, Umwelt und Frauen Bildungsraum GrünGürtel "Im Bewußtsein ihrer Verantwortung für die nachfolgende Generation" beschloss die Stadtverordnetenversammlung der Stadt Frankfurt 1991, die Landschaft rings um die Kernstadt als "GrünGürtel" zum Wohl von Mensch und Natur zu sichern und weiterzuentwickeln. Diese Zielsetzung beinhaltet, Kindern und Jugendlichen Wert und Bedeutung des Einleitung GrünGürtel zu vermitteln, um für die Zukunft den Bestand und die nach- Projekte haltige Entwicklung der Grünräume zu gewährleisten. Die hiermit im Spielorte Zusammenhang stehenden Themen und Fragestellungen des GrünGür- Lernstationen tels sind bislang noch nicht Gegenstand von konzeptionellen Überlegun- Fahrradprojekt gen für die pädagogische Arbeit in Kindertagesstätten und Schulen. Der Kooperationspartner vorliegende Entwurf versteht sich als Beitrag, den GrünGürtel als Bil- Projektplan dungsraum für Kinder und Jugendliche zu entwickeln. Literatur Für Kinder bieten die Landschaftsräume des GrünGürtels vielfältige Erlebnis- und Erfahrungsmöglichkeiten bei Sport und Spiel, beim Stro- mern und Durchstreifen der Natur, aus denen eine emotionale Bindung zu diesen Räumen erwachsen kann. Das Angebot der GrünGürtel-Land- schaft nehmen Kinder heute allerdings nur eingeschränkt wahr, da das Sicherheitsempfinden von Eltern, die erschwerte Zugänglichkeit und das veränderte Freizeitverhalten von Kindern und Jugendlichen einer indivi- duellen Aneignung der Naturräume -

Nieder-Eschbach Bonames Riedberg Heddernheim
Nieder-Eschbach Bonames Riedberg N8 Heddernheim Eschersheimer Landstr. Südbahnhof RMV-Servicetelefon: 069 / 24 24 80 24 ▼ Das Fahrtenangebot in den Wochenendnächten finden Sie in der U-Bahn-Linie U8. Nieder-EschbachOtto-Hahn-SchuleWilhelm-Flögel-RingNieder-EschbachAn der BornhohlKonrad-Duden-WegBonames MitteBonameserKalbach HainstraßeBonifatiusstraßeAlte RiedbergstraßeSchule KalbachRathaus KalbachAm WeißkirchenerSchwalbenweg BergPaul-Apel-StraßeAnnette-Kolb-WegLudwig-Fulda-WegRiedberg Käthe-Kruse-Straße 662 29 29 28 Am BonifatiusbrunnenOberschelderHeddernheim Weg Weißer SteinLindenbaumHügelstraßeFritz-Tarnow-StraßeDornbusch/AmDornbusch GrünhofMiquel-/AdickesalleeHolzhausenstraßeGrüneburgwegEschenheimerFreßgass/Hauptwache Tor Roßmarkt Freßgass TaunusanlageWilly-Brandt-PlatzSchwanthalerstraße Südbahnhof M60 M34 M34 M32 M36 N7 S-Bahn S-Bahn N4 N11 N16 Regional- S-Bahn N16 M36 N5 N12 und Fern- 61 verkehr Nächte So/Mo bis Do/Fr, nicht vor Feiertagen 29 Hohe Brück ab 1.15 2.15 3.10 29 Nieder-Eschbach an 1.24 2.24 3.19 Hinweise 29> 29> 29> Nieder-Eschbach [U] # 0.53 # 1.53 # 2.53 Nieder-Eschbach [U] ★ 0.54 ★ 1.24 ★ 1.54 ★ 2.24 ★ 2.54 ★ 3.19 An der Bornhohl 0.55 1.25 1.55 2.25 2.55 3.20 Konrad-Duden-Weg 0.57 1.27 1.57 2.27 2.57 3.22 Bonames Mitte 0.59 1.29 1.59 2.29 2.59 3.24 Bonameser Hainstraße 1.00 1.30 2.00 2.30 3.00 3.25 Kalbach [U] 1.02 1.32 2.02 2.32 3.02 3.27 Alte Riedbergstraße 1.04 1.34 2.04 2.34 3.04 3.29 Schule Kalbach 1.06 1.36 2.06 2.36 3.06 3.31 Rathaus Kalbach 1.07 1.37 2.07 2.37 3.07 3.32 Am Weißkirchener Berg 1.08 1.38 2.08 -

Office Market Profile
Office Market Profile Frankfurt | 1st quarter 2020 Published in April 2020 Frankfurt Development of Main Indicators Low space take-up in the fi rst quarter Take-up in the fi rst quarter of 2020 was just 67,600 sqm, the fourth worst quarterly take-up result for more than 20 years. This was mainly due to the low deal overhang from the previous year, as the fourth quarter of 2019 was the third strongest quarter in the last ten years, and to the eff ects of the current coronavirus crisis. In contrast, the number of deals was comparatively high, since mainly medium and large-sized searches for space were post- poned. The vacancy rate rose slightly to 5.8% due to infrastructure; these investments will also have an impact numerous vacancies resulting from contracts signed in on space concepts in the medium and long-term. If the 2018 and 2019. Across the submarkets, prime and average catch-up eff ects come in the second half of the year, over- rents remained unchanged. In the current economic all performance in 2020 could remain strong, provided climate, owners and (existing) tenants are engaged in that there is enough time for deals to be concluded and intensive exchanges and solutions are being sought in that property owners do not put together too attractive the form of incentives, subletting or fl exible contractual lease extension packages. arrangements. In the short term, many companies will focus on creating or improving their remote working Frankfurt: Off ice Space Market Areas with Rental Bands (€/sqm/month) Frankfurt: Office Space Market Areas with Rental Bands (€/sqm/month) JLL Research 2020/Q1 Kalbach Oberhoechstadt/Ts.