Parkinson's Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
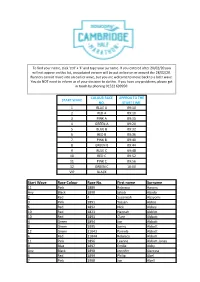
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Surnames 198
Surnames 198 P PACQUIN PAGONE PALCISCO PACUCH PAHACH PALEK PAAHANA PACY PAHEL PALENIK PAAR PADASAK PAHUSZKI PALERMO PAASSARELLI PADDOCK PAHUTSKY PALESCH PABALAN PADELL PAINE PALGUTA PABLIK PADGETT PAINTER PALI PABRAZINSKY PADLO PAIRSON PALILLA PABST PADUNCIC PAISELL PALINA PACCONI PAESANI PAJAK PALINO PACE PAESANO PAJEWSKI PALINSKI PACEK PAFFRATH PAKALA PALKO PACELLI PAGANI PAKOS PALL PACEY PAGANO PALACE PALLO PACHARKA PAGDEN PALADINO PALLONE PACIFIC PAGE PALAGGO PALLOSKY PACILLA PAGLARINI PALAIC PALLOTTINI PACINI PAGLIARINI PALANIK PALLOZZI PACK PAGLIARNI PALANKEY PALM PACKARD PAGLIARO PALANKI PALMA PACKER PAGLIARULO PALAZZONE PALMER PACNUCCI PAGLIASOTTI PALCHESKO PALMERO PACOLT PAGO PALCIC PALMERRI Historical & Genealogical Society of Indiana County 1/21/2013 Surnames 199 PALMIERI PANCIERRA PAOLO PARDUS PALMISANO PANCOAST PAONE PARE PALMISCIANO PANCZAK PAPAKIE PARENTE PALMISCNO PANDAL PAPCIAK PARENTI PALMO PANDULLO PAPE PARETTI PALOMBO PANE PAPIK PARETTO PALONE PANGALLO PAPOVICH PARFITT PALSGROVE PANGBURN PAPPAL PARHAM PALUCH PANGONIS PAPSON PARILLO PALUCHAK PANIALE PAPUGA PARIS PALUDA PANKOVICH PAPURELLO PARISE PALUGA PANKRATZ PARADA PARISEY PALUGNACK PANNACHIA PARANA PARISH PALUMBO PANNEBAKER PARANIC PARISI PALUS PANONE PARAPOT PARISO PALUSKA PANOSKY PARATTO PARIZACK PALYA PANTALL PARCELL PARK PAMPE PANTALONE PARCHINSKY PARKE PANAIA PANTANI PARCHUKE PARKER PANASCI PANTANO PARDEE PARKES PANASKI PANTZER PARDINI PARKHILL PANCHICK PANZY PARDO PARKHURST PANCHIK PAOLINELLIE PARDOE PARKIN Historical & Genealogical Society of Indiana County -

British Family Names
cs 25o/ £22, Cornrll IBniwwitg |fta*g BOUGHT WITH THE INCOME FROM THE SAGE ENDOWMENT FUND THE GIFT OF Hcnrti W~ Sage 1891 A.+.xas.Q7- B^llll^_ DATE DUE ,•-? AUG 1 5 1944 !Hak 1 3 1^46 Dec? '47T Jan 5' 48 ft e Univeral, CS2501 .B23 " v Llb«"y Brit mii!Sm?nS,£& ori8'" and m 3 1924 olin 029 805 771 The original of this book is in the Cornell University Library. There are no known copyright restrictions in the United States on the use of the text. http://www.archive.org/details/cu31924029805771 BRITISH FAMILY NAMES. : BRITISH FAMILY NAMES ftbetr ©riain ano fIDeaning, Lists of Scandinavian, Frisian, Anglo-Saxon, and Norman Names. HENRY BARBER, M.D. (Clerk), "*• AUTHOR OF : ' FURNESS AND CARTMEL NOTES,' THE CISTERCIAN ABBEY OF MAULBRONN,' ( SOME QUEER NAMES,' ' THE SHRINE OF ST. BONIFACE AT FULDA,' 'POPULAR AMUSEMENTS IN GERMANY,' ETC. ' "What's in a name ? —Romeo and yuliet. ' I believe now, there is some secret power and virtue in a name.' Burton's Anatomy ofMelancholy. LONDON ELLIOT STOCK, 62, PATERNOSTER ROW, E.C. 1894. 4136 CONTENTS. Preface - vii Books Consulted - ix Introduction i British Surnames - 3 nicknames 7 clan or tribal names 8 place-names - ii official names 12 trade names 12 christian names 1 foreign names 1 foundling names 1 Lists of Ancient Patronymics : old norse personal names 1 frisian personal and family names 3 names of persons entered in domesday book as HOLDING LANDS temp. KING ED. CONFR. 37 names of tenants in chief in domesday book 5 names of under-tenants of lands at the time of the domesday survey 56 Norman Names 66 Alphabetical List of British Surnames 78 Appendix 233 PREFACE. -

Most-Common-Surnames-Bmd-Registers-16.Pdf
Most Common Surnames Surnames occurring most often in Scotland's registers of Births, Marriages and Deaths Counting only the surname of the child for births, the surnames of BOTH PARTIES (for example both BRIDE and GROOM) for marriages, and the surname of the deceased for deaths Note: the surnames from these registers may not be representative of the surnames of the population of Scotland as a whole, as (a) they include the surnames of non-residents who were born / married / died here; (b) they exclude the surnames of residents who were born / married / died elsewhere; and (c) some age-groups have very low birth, marriage and death rates; others account for most births, marriages and deaths.ths Registration Year = 2016 Position Surname Number 1 SMITH 2056 2 BROWN 1435 3 WILSON 1354 4 CAMPBELL 1147 5 STEWART 1139 6 THOMSON 1127 7 ROBERTSON 1088 8 ANDERSON 1001 9 MACDONALD 808 10 TAYLOR 782 11 SCOTT 771 12 REID 755 13 MURRAY 754 14 CLARK 734 15 WATSON 642 16 ROSS 629 17 YOUNG 608 18 MITCHELL 601 19 WALKER 589 20= MORRISON 587 20= PATERSON 587 22 GRAHAM 569 23 HAMILTON 541 24 FRASER 529 25 MARTIN 528 26 GRAY 523 27 HENDERSON 522 28 KERR 521 29 MCDONALD 520 30 FERGUSON 513 31 MILLER 511 32 CAMERON 510 33= DAVIDSON 506 33= JOHNSTON 506 35 BELL 483 36 KELLY 478 37 DUNCAN 473 38 HUNTER 450 39 SIMPSON 438 40 MACLEOD 435 41 MACKENZIE 434 42 ALLAN 432 43 GRANT 429 44 WALLACE 401 45 BLACK 399 © Crown Copyright 2017 46 RUSSELL 394 47 JONES 392 48 MACKAY 372 49= MARSHALL 370 49= SUTHERLAND 370 51 WRIGHT 357 52 GIBSON 356 53 BURNS 353 54= KENNEDY 347 -

Hebron Maple Festival This Weekend RHAM Juniors Hold Fashion Show
★ ★ ★ ★ ★ US. POSTAGE PAID POSTAL CUSTOMER GLASTONBURY CITIZEN, INC. LOCAL RIVEREAST PRESORTED STANDARD NewsServing Amston, Andover, Cobalt, East Hampton, Hebron,Bulletin Marlborough, Middle Haddam, Portland, Colchester and Salem Volume 33, Number 51 Published by The Glastonbury Citizen March 13, 2009 Hebron Maple Festival This Weekend by Sarah McCoy The steel buckets hanging from the sides of explained that the ideal syrup-making weather trees can only mean one thing. With spring just features nighttime temperatures in the low 20s, around the corner, the time has come for the with daytime highs in the 40s, with lots of sun 19th annual Hebron Maple Festival. and little to no wind. While this year’s season The festival will be held this year Saturday started off terribly, both sugarers agreed that and Sunday, March 14 and 15, from 10 a.m.-4 the past few weeks have made up for the slow p.m. both days. The affair will feature events start. “This year might prove to be average,” across the town and plenty of food. Palmer said. “That would be a huge achieve- “It’s unique,” Wayne Palmer, owner of ment considering how the season started.” Winding Brook Sugar House and one of the Palmer places taps in over 850 trees. Last coordinators for this year’s festival, said. year, he said, he produced 250 gallons of syrup. “We’re the only town in Connecticut that does “Last year was one of the best in history,” a maple festival and I’ve been told it rivals any- Palmer said. “It was an anomaly but we could thing in Vermont because we’ve kept money get to 200 this year.” out of the equation.” In addition to its use on pancakes and As in years past, there is free admission to waffles, syrup can be used for all different rea- the Maple Festival. -

2006 Kyoto, Japan
October 28 - November 2, 2006 ~ Kyoto, Japan ~ Final Program Table of Contents Welcome . .2 Acknowledgements . .3 Organization . .6 MDS .Committees .& Task. .Forces . .9 International .Congress .Registration .and Venue. .12 International .Congress .Information . 13-15 . Continuing .Medical .Education . .13 . Evaluations . .14 . Press .Room . .15 Program-at-a-Glance . .17 Scientific Session Definitions . .19 Scientific .Sessions . .21 Faculty . .51 Committee .& Task. .Force .Meetings . .56 Exhibitor .Information . .57 Exhibitor .Directory . .58 Floor .Plans . 62-64 Map .of .Kyoto . .66 Lunch Map . .67 Subway Map . .68 Social Events . .69 Poster .Session .1 . .72 Poster .Session .2 . .88 Poster .Session .3 . .102 Poster .Session .4 . .117 CME .Request .Form . .133 The Movement. .Disorder .Society’s 0th International Congress of Parkinson’s Disease and Movement Disorders Welcome Letter Dear Colleagues, On behalf of The Movement Disorder Society (MDS), we are pleased to welcome you to Kyoto, Japan for the 10th International Congress of Parkinson’s Disease and Movement Disorders . The 10th International Congress has been designed to provide an innovative and comprehensive overview of the latest perspectives and research developments in the field of Movement Disorders . We encourage you to take every opportunity to participate in the Scientific Program which has drawn world renowned speakers and foremost experts in their respective fields . In the next days, the latest research regarding Movement Disorders will be presented and discussed in an open format, offering unique educational opportunities for all delegates . The International Congress convenes with a series of Opening Seminars and then continues with an array of Plenary, Parallel, Poster and Video Sessions, as well as Lunch Seminars, Controversies and Skills Workshops . -

Iain Sinclair
Iain Sinclair: An Inventory of His Papers at the Harry Ransom Center Descriptive Summary Creator: Sinclair, Iain, 1943- Title: Iain Sinclair Papers Dates: 1882-2009 (bulk 1960s-2008) Extent: 135 document boxes, 8 oversize boxes (osb) (56.7 linear feet), 23 oversize folders (osf), 15 computer disks Abstract: The papers of British writer Iain Sinclair consist of drafts of works, research material, juvenilia, notebooks, personal and professional correspondence, business files, financial files, works by others, ephemera, and electronic files. They document Sinclair’s prolific and diverse career, from running his own press to his wide range of creative output including works of poetry, fiction, non-fiction, edited anthologies, screenplays, articles, essays, reviews, and radio and television contributions. Call Number: Manuscript Collection MS-4930 Language: English; some French, German, and Italian Access: Open for research. Researchers must register and agree to copyright and privacy laws before using archival materials. Some materials restricted due to condition and conservation status. Use Policies: Ransom Center collections may contain material with sensitive or confidential information that is protected under federal or state right to privacy laws and regulations. Researchers are advised that the disclosure of certain information pertaining to identifiable living individuals represented in the collections without the consent of those individuals may have legal ramifications (e.g., a cause of action under common law for invasion of privacy may arise if facts concerning an individual's private life are published that would be deemed highly offensive to a reasonable person) for which the Ransom Center and The University of Texas at Austin assume no responsibility. -

ASSESSED VALUE REVISIONS Downers Grove Township, 2019
NOTICE TO TAXPAYERS – ASSESSED VALUE REVISIONS Downers Grove Township, 2019 Assessed Values Median Level of Assessment: 33 1/3% Valuation Date: 01/01/2019 Relevant Real Estate Sales Information Used to Develop the Equalized Assessed Value: Sales Occurring Between 01/01/2016 – 12/31/2018 Your property is to be assessed at the above listed median level of assessment for the assessment district. You may check the accuracy of your assessment by dividing your assessment by the median level of assessment. The resulting value should equal the estimated fair cash value of your property. If the resulting value is greater than the estimated fair cash value of your property, you may be over-assessed. If the resulting value is less than the fair cash value of your property, you may be under-assessed. You may appeal your assessment to the Board of Review Illinois law requires assessed values of property, other than farmland and coal, are to be assessed of 33 1/3% of fair market value. Restrictions within Illinois law delay the change in property assessments caused by changes of actual property fair cash value. State law requires the assessed values are to be adjusted based upon data from the three prior calendar years before the assessment date. In appreciating markets, this forces current property assessments to lag behind recent sales prices, and in declining markets, the decline of assessed values is delayed. Contact your local assessor’s office to review your assessment if you believe the fair cash value is incorrect or you are not assessed uniformly with comparable properties in your neighborhood. -

Obituary Index
Yamhill County Obituary Index Surnames:M-Z Obituaries contained in the files at the Yamhill County Historical Society Research Library in Lafayette, Oregon 1850s through October 2019 Yamhill County Historical Society 657 Market Street Lafayette, Oregon 97127 www.yamhillcountyhistory.org Obituary clippings are pasted to note cards and filed alphabetically. Copies can be obtained by contacting us at by phone or email, or in person during our open hours. Current contact information can be found at www.yamhillcountyhistory.org A duplication fee will be incurred for copies made. Obituaries have been gathered from current and discontinued newspapers Local Newspapers Include: • News Register (McMinnville) • Newberg Graphic • Sheridan Sun Regional Newspapers • Oregonian (Portland) • Statesman Journal (Salem) Maahs, Faythe Dec 25, 1999 Maahs, Leonard H Apr 12, 2003 Maas, Agnes “Peggy” May 12, 1975 Maas, Catharina Wilhelmina Aug 22, 2010 Maas, Edna E Jan 20, 1981 Maas, Jacob B Apr 2, 1984 Maas, Jerry B Aug 27, 2005 Maas, Petrus G “Peter” Jul 24, 2002 Mabal, Linnie Bewley Feb 16, 1930 Mabee, Donald F Nov 14, 1996 Mabee, Matilda Mar 8, 1937 Mabee, Susan Bernice Apr 13, 2017 Maben, Richard Lee Mar 24, 2018 Maben, Robert “Bob” May 20, 2019 Mabry, Doris A Nov 17, 2005 Mabry, Jack Roger Jul 26, 2012 Mabry, Laura Belle Mar 23, 2018 Mabry, Mildred R Jan 2, 2012 MacArthur, Donald F Dec 8, 2009 Macauley, Donna Lynn Nov 12, 1989 Macauley, John G Nov 6, 1974 Macauley, Margaret Jul 13, 2010 Macauley, Murray “Neil” Nov 12, 2008 MacBeth, Gordon Dec 10, 2014 MacBeth, -
Iron, Aging, and Neurodegeneration
Metals 2015, 5, 2070-2092; doi:10.3390/met5042070 OPEN ACCESS metals ISSN 2075-4701 www.mdpi.com/journal/metals/ Review Iron, Aging, and Neurodegeneration Dafina M. Angelova and David R. Brown * Department of Biology and Biochemistry, University of Bath, Bath BA2 7AY, UK; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +44-1225-383133; Fax: +44-1225-386779. Academic Editor: Grasso Giuseppe Received: 5 October 2015 / Accepted: 2 November 2015 / Published: 6 November 2015 Abstract: Iron is a trace element of considerable interest to both chemistry and biology. In a biological context its chemistry is vital to the roles it performs. However, that same chemistry can contribute to a more deleterious role in a variety of diseases. The brain is a very sensitive organ due to the irreplaceable nature of neurons. In this regard regulation of brain iron chemistry is essential to maintaining neuronal viability. During the course of normal aging, the brain changes the way it deals with iron and this can contribute to its susceptibility to disease. Additionally, many of the known neurodegenerative diseases have been shown to be influenced by changes in brain iron. This review examines the role of iron in the brain and neurodegenerative diseases and the potential role of changes in brain iron caused by aging. Keywords: synuclein; amyloid; prion; Alzheimer’s disease; Parkinson’s disease; transmissible spongiform encephalopathy; ferrireductase; microglia 1. Introduction While the atomic nature of matter might be an absolute given in the 21st century, the role of metal atoms in biological systems remains a developing field. -
Index to St. Louis, Missouri Naturalization Records Created After Sept
Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 Alphabetical surname index N–R History & Genealogy Department St. Louis County Library 1640 S. Lindberg Blvd. St. Louis, Missouri 63131 314-994-3300, ext. 2070 [email protected] Index to St. Louis, Missouri Naturalization Records Created after Sept. 27, 1906 This index covers St. Louis, Missouri naturalization records created between October 1, 1906 and December 1928 and is based on the following sources: • Naturalizations, U.S. District Court—Eastern Division, Eastern Judicial District of Missouri, Vols. 1 – 82 • Naturalizations, U.S. Circuit Court— Eastern Division, Eastern Judicial District of Missouri, Vols. 5 – 21 Entries are listed alphabetically by surname, then by given name, and then numerically by volume number. Abbreviations and Notations SLCL = History and Genealogy Department microfilm number (St. Louis County Library) FHL = Family History Library microfilm number * = spelling taken from the signature which differed from name in index. How to obtain copies Photocopies of indexed articles may be requested by sending an email to the History and Genealogy Department at [email protected]. A limit of three searches per request applies. Please review the library's lookup policy at https://www.slcl.org/genealogy-and-local- history/services. A declaration of intention may lead to further records. For more information, contact the National Archives at the address below. Include all information listed on the declaration of intention. National Archives, Central Plains Region 400 W. Pershing Rd. Kansas City, MO 64108 (816) 268-8000 [email protected] History Genealogy Dept. Index to St. Louis, Missouri Naturalization Records St. -

REFERENCES to English Surnames in 1602
REFERENCES TO ENGLISH SURNAMES 1602. cs 2505 .H62 1911 DUKE UNIVERSITY LIBRARY Digitized by the Internet Archive in 2018 with funding from Duke University Libraries https://archive.org/details/referencestoengl01 hitc_0 REFERENCES TO English Surnames IN 1602 AN INDEX GIVING ABOUT 20,500 REFERENCES TO SURNAMES CONTAINED IN THE PRINTED REGISTERS OF 964 ENGLISH PARISHES DURING THE SECOND YEAR OF THE XVII. CENTURY. WITH AN APPENDIX INDEXING THE SURNAMES CONTAINED IN 186 PRINTED REGISTERS DURING 1601 (OMITTED FROM THE VOLUME FOR THAT YEAR) BY F. K. & S. HITCHING. 1911 CHAS. A. BERNAU, 17. BILLITER SQUARE BUILDINGS. LONDON. THE ST. CATHERINE PRESS 34, NORFOLK STREET STRAND, W.C. HtelSKC PREFACE TO THE VOLUME FOR j6oj (Reprinted) After even fifty years’ original research among our old records, a genealogist, interested in the history of all the branches of one English family, would know that many branches remained to be traced. Much of his time would have been wasted in searching “ likely ” registers, which, on examination, proved to contain no data for him. Experience teaches every earnest student of family history that what he needs, is not more records, but more clues as to the contents of the available records. “ Indexes and more Indexes ” is his cry, and the publisher who issues a genealogical work without an index merits (and often receives) his imprecations. No records require an Index so urgently as the Parish Registers, which record the baptisms, marriages and burials of our ancestors. These are spread over all the country, and, not only is there no general index to their contents, but there is not even an Index in each Parish to its own Registers.