Iron, Aging, and Neurodegeneration
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Parkinson's Disease and Metal Storage Disorders
brain sciences Review Parkinson’s Disease and Metal Storage Disorders: A Systematic Review Edward Botsford 1,* , Jayan George 2,3 and Ellen E. Buckley 4,5 1 University of Sheffield Medical School, Beech Hill Road, Sheffield S10 2RX, UK 2 General Surgical Department, Sheffield Teaching Hospitals NHS Foundation Trust, Herries Road, Sheffield S5 7AU, UK; [email protected] 3 University of Sheffield, Western Bank, S10 2TN Sheffield, UK 4 Sheffield Institute for Translational Neuroscience, University of Sheffield, 385a Glossop Road, Sheffield S10 2HQ, UK; e.e.buckley@sheffield.ac.uk 5 INSIGNEO Institute for in silico Medicine, University of Sheffield, Pam Liversidge Building, Sheffield S1 3JD, UK * Correspondence: ebotsford1@sheffield.ac.uk; Tel.: +44-(0)114-222-2272 Received: 9 October 2018; Accepted: 30 October 2018; Published: 31 October 2018 Abstract: Metal storage disorders (MSDs) are a set of rare inherited conditions with variable clinical pictures including neurological dysfunction. The objective of this study was, through a systematic review, to identify the prevalence of Parkinsonism in patients with MSDs in order to uncover novel pathways implemented in Parkinson’s disease. Human studies describing patients of any age with an MSD diagnosis were analysed. Foreign language publications as well as animal and cellular studies were excluded. Searches were conducted through PubMed and Ovid between April and September 2018. A total of 53 publications were identified including 43 case reports, nine cross-sectional studies, and one cohort study. The publication year ranged from 1981 to 2018. The most frequently identified MSDs were Pantothenate kinase-associated neurodegeneration (PKAN) with 11 papers describing Parkinsonism, Hereditary hemochromatosis (HH) (7 papers), and Wilson’s disease (6 papers). -
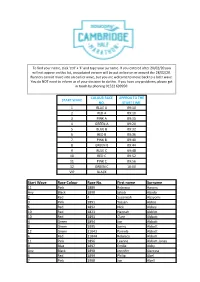
Start Wave Race Colour Race No. First Name Surname
To find your name, click 'ctrl' + 'F' and type your surname. If you entered after 20/02/20 you will not appear on this list, an updated version will be put online on or around the 28/02/20. Runners cannot move into an earlier wave, but you are welcome to move back to a later wave. You do NOT need to inform us of your decision to do this. If you have any problems, please get in touch by phoning 01522 699950. COLOUR RACE APPROX TO THE START WAVE NO. START TIME 1 BLUE A 09:10 2 RED A 09:10 3 PINK A 09:15 4 GREEN A 09:20 5 BLUE B 09:32 6 RED B 09:36 7 PINK B 09:40 8 GREEN B 09:44 9 BLUE C 09:48 10 RED C 09:52 11 PINK C 09:56 12 GREEN C 10:00 VIP BLACK Start Wave Race Colour Race No. First name Surname 11 Pink 1889 Rebecca Aarons Any Black 1890 Jakob Abada 2 Red 4 Susannah Abayomi 3 Pink 1891 Yassen Abbas 6 Red 1892 Nick Abbey 10 Red 1823 Hannah Abblitt 10 Red 1893 Clare Abbott 4 Green 1894 Jon Abbott 8 Green 1895 Jonny Abbott 12 Green 11043 Pamela Abbott 6 Red 11044 Rebecca Abbott 11 Pink 1896 Leanne Abbott-Jones 9 Blue 1897 Emilie Abby Any Black 1898 Jennifer Abecina 6 Red 1899 Philip Abel 7 Pink 1900 Jon Abell 10 Red 600 Kirsty Aberdein 6 Red 11045 Andrew Abery Any Black 1901 Erwann ABIVEN 11 Pink 1902 marie joan ablat 8 Green 1903 Teresa Ablewhite 9 Blue 1904 Ahid Abood 6 Red 1905 Alvin Abraham 9 Blue 1906 Deborah Abraham 6 Red 1907 Sophie Abraham 1 Blue 11046 Mitchell Abrams 4 Green 1908 David Abreu 11 Pink 11047 Kathleen Abuda 10 Red 11048 Annalisa Accascina 4 Green 1909 Luis Acedo 10 Red 11049 Vikas Acharya 11 Pink 11050 Catriona Ackermann -

Surnames 198
Surnames 198 P PACQUIN PAGONE PALCISCO PACUCH PAHACH PALEK PAAHANA PACY PAHEL PALENIK PAAR PADASAK PAHUSZKI PALERMO PAASSARELLI PADDOCK PAHUTSKY PALESCH PABALAN PADELL PAINE PALGUTA PABLIK PADGETT PAINTER PALI PABRAZINSKY PADLO PAIRSON PALILLA PABST PADUNCIC PAISELL PALINA PACCONI PAESANI PAJAK PALINO PACE PAESANO PAJEWSKI PALINSKI PACEK PAFFRATH PAKALA PALKO PACELLI PAGANI PAKOS PALL PACEY PAGANO PALACE PALLO PACHARKA PAGDEN PALADINO PALLONE PACIFIC PAGE PALAGGO PALLOSKY PACILLA PAGLARINI PALAIC PALLOTTINI PACINI PAGLIARINI PALANIK PALLOZZI PACK PAGLIARNI PALANKEY PALM PACKARD PAGLIARO PALANKI PALMA PACKER PAGLIARULO PALAZZONE PALMER PACNUCCI PAGLIASOTTI PALCHESKO PALMERO PACOLT PAGO PALCIC PALMERRI Historical & Genealogical Society of Indiana County 1/21/2013 Surnames 199 PALMIERI PANCIERRA PAOLO PARDUS PALMISANO PANCOAST PAONE PARE PALMISCIANO PANCZAK PAPAKIE PARENTE PALMISCNO PANDAL PAPCIAK PARENTI PALMO PANDULLO PAPE PARETTI PALOMBO PANE PAPIK PARETTO PALONE PANGALLO PAPOVICH PARFITT PALSGROVE PANGBURN PAPPAL PARHAM PALUCH PANGONIS PAPSON PARILLO PALUCHAK PANIALE PAPUGA PARIS PALUDA PANKOVICH PAPURELLO PARISE PALUGA PANKRATZ PARADA PARISEY PALUGNACK PANNACHIA PARANA PARISH PALUMBO PANNEBAKER PARANIC PARISI PALUS PANONE PARAPOT PARISO PALUSKA PANOSKY PARATTO PARIZACK PALYA PANTALL PARCELL PARK PAMPE PANTALONE PARCHINSKY PARKE PANAIA PANTANI PARCHUKE PARKER PANASCI PANTANO PARDEE PARKES PANASKI PANTZER PARDINI PARKHILL PANCHICK PANZY PARDO PARKHURST PANCHIK PAOLINELLIE PARDOE PARKIN Historical & Genealogical Society of Indiana County -

A Pilot Trial of Deferiprone for Neurodegeneration with Brain Iron Accumulation
BRIEF REPORTS A pilot trial of deferiprone for neurodegeneration with brain iron accumulation Giovanni Abbruzzese, 1 Giovanni Cossu, 2 Manuela Balocco, 3 Roberta Marchese, 1 Daniela Murgia, 2 Maurizio Melis, 2 Renzo Galanello, 4 Susanna Barella, 4 Gildo Matta, 5 Uberto Ruffinengo, 6 Ubaldo Bonuccelli, 7 and Gian Luca Forni, 3 1Department of Neurosciences, University of Genoa; 2Neurology Department, “G. Brotzu” General Hospital, Cagliari; 3Centro della Microcitemia e Anemie Congenite-Haematology, Galliera Hospital, Genoa; 4Ospedale Regionale Microcitemia, University of Cagliari, Cagliari; 5Radiology Department “G. Brotzu” General Hospital, Cagliari; 6Neuroradiology Unit, Galliera Hospital, Genoa; 7Neuroscience Department, University of Pisa, Italy ABSTRACT Deferiprone was shown to reverse iron deposition in associated neurodegeneration). These results underline the Friedreich's ataxia. This multi-center, unblinded, single-arm safety and tolerability of deferiprone, and suggest that pilot study evaluated safety and efficacy of deferiprone for chelating treatment might be effective in improving neuro - reducing cerebral iron accumulation in neurodegeneration logical manifestations associated with iron accumulation. with brain iron accumulation. Four patients with genetical - (Clinicaltrials.gov Identifier: NTC00907283) ly-confirmed pantothenate kinase-associated neurodegener - ation, and 2 with parkinsonism and focal dystonia, but Key words: deferiprone, iron overload, neurodegeneration inconclusive genetic tests, received 15 mg/kg deferiprone with brain iron accumulation. bid. Magnetic resonance imaging and neurological examina - tions were conducted at baseline, six and 12 months. Citation: Abbruzzese G, Cossu G, Balocco M, Marchese R, Chelation treatment caused no apparent hematologic or Murgia D, Melis M, Galanello R, Barella S, Matta G, neurological side effects. Magnetic resonance imaging Ruffinengo U, Bonuccelli U, and Forni GL. -

British Family Names
cs 25o/ £22, Cornrll IBniwwitg |fta*g BOUGHT WITH THE INCOME FROM THE SAGE ENDOWMENT FUND THE GIFT OF Hcnrti W~ Sage 1891 A.+.xas.Q7- B^llll^_ DATE DUE ,•-? AUG 1 5 1944 !Hak 1 3 1^46 Dec? '47T Jan 5' 48 ft e Univeral, CS2501 .B23 " v Llb«"y Brit mii!Sm?nS,£& ori8'" and m 3 1924 olin 029 805 771 The original of this book is in the Cornell University Library. There are no known copyright restrictions in the United States on the use of the text. http://www.archive.org/details/cu31924029805771 BRITISH FAMILY NAMES. : BRITISH FAMILY NAMES ftbetr ©riain ano fIDeaning, Lists of Scandinavian, Frisian, Anglo-Saxon, and Norman Names. HENRY BARBER, M.D. (Clerk), "*• AUTHOR OF : ' FURNESS AND CARTMEL NOTES,' THE CISTERCIAN ABBEY OF MAULBRONN,' ( SOME QUEER NAMES,' ' THE SHRINE OF ST. BONIFACE AT FULDA,' 'POPULAR AMUSEMENTS IN GERMANY,' ETC. ' "What's in a name ? —Romeo and yuliet. ' I believe now, there is some secret power and virtue in a name.' Burton's Anatomy ofMelancholy. LONDON ELLIOT STOCK, 62, PATERNOSTER ROW, E.C. 1894. 4136 CONTENTS. Preface - vii Books Consulted - ix Introduction i British Surnames - 3 nicknames 7 clan or tribal names 8 place-names - ii official names 12 trade names 12 christian names 1 foreign names 1 foundling names 1 Lists of Ancient Patronymics : old norse personal names 1 frisian personal and family names 3 names of persons entered in domesday book as HOLDING LANDS temp. KING ED. CONFR. 37 names of tenants in chief in domesday book 5 names of under-tenants of lands at the time of the domesday survey 56 Norman Names 66 Alphabetical List of British Surnames 78 Appendix 233 PREFACE. -

Most-Common-Surnames-Bmd-Registers-16.Pdf
Most Common Surnames Surnames occurring most often in Scotland's registers of Births, Marriages and Deaths Counting only the surname of the child for births, the surnames of BOTH PARTIES (for example both BRIDE and GROOM) for marriages, and the surname of the deceased for deaths Note: the surnames from these registers may not be representative of the surnames of the population of Scotland as a whole, as (a) they include the surnames of non-residents who were born / married / died here; (b) they exclude the surnames of residents who were born / married / died elsewhere; and (c) some age-groups have very low birth, marriage and death rates; others account for most births, marriages and deaths.ths Registration Year = 2016 Position Surname Number 1 SMITH 2056 2 BROWN 1435 3 WILSON 1354 4 CAMPBELL 1147 5 STEWART 1139 6 THOMSON 1127 7 ROBERTSON 1088 8 ANDERSON 1001 9 MACDONALD 808 10 TAYLOR 782 11 SCOTT 771 12 REID 755 13 MURRAY 754 14 CLARK 734 15 WATSON 642 16 ROSS 629 17 YOUNG 608 18 MITCHELL 601 19 WALKER 589 20= MORRISON 587 20= PATERSON 587 22 GRAHAM 569 23 HAMILTON 541 24 FRASER 529 25 MARTIN 528 26 GRAY 523 27 HENDERSON 522 28 KERR 521 29 MCDONALD 520 30 FERGUSON 513 31 MILLER 511 32 CAMERON 510 33= DAVIDSON 506 33= JOHNSTON 506 35 BELL 483 36 KELLY 478 37 DUNCAN 473 38 HUNTER 450 39 SIMPSON 438 40 MACLEOD 435 41 MACKENZIE 434 42 ALLAN 432 43 GRANT 429 44 WALLACE 401 45 BLACK 399 © Crown Copyright 2017 46 RUSSELL 394 47 JONES 392 48 MACKAY 372 49= MARSHALL 370 49= SUTHERLAND 370 51 WRIGHT 357 52 GIBSON 356 53 BURNS 353 54= KENNEDY 347 -

Hebron Maple Festival This Weekend RHAM Juniors Hold Fashion Show
★ ★ ★ ★ ★ US. POSTAGE PAID POSTAL CUSTOMER GLASTONBURY CITIZEN, INC. LOCAL RIVEREAST PRESORTED STANDARD NewsServing Amston, Andover, Cobalt, East Hampton, Hebron,Bulletin Marlborough, Middle Haddam, Portland, Colchester and Salem Volume 33, Number 51 Published by The Glastonbury Citizen March 13, 2009 Hebron Maple Festival This Weekend by Sarah McCoy The steel buckets hanging from the sides of explained that the ideal syrup-making weather trees can only mean one thing. With spring just features nighttime temperatures in the low 20s, around the corner, the time has come for the with daytime highs in the 40s, with lots of sun 19th annual Hebron Maple Festival. and little to no wind. While this year’s season The festival will be held this year Saturday started off terribly, both sugarers agreed that and Sunday, March 14 and 15, from 10 a.m.-4 the past few weeks have made up for the slow p.m. both days. The affair will feature events start. “This year might prove to be average,” across the town and plenty of food. Palmer said. “That would be a huge achieve- “It’s unique,” Wayne Palmer, owner of ment considering how the season started.” Winding Brook Sugar House and one of the Palmer places taps in over 850 trees. Last coordinators for this year’s festival, said. year, he said, he produced 250 gallons of syrup. “We’re the only town in Connecticut that does “Last year was one of the best in history,” a maple festival and I’ve been told it rivals any- Palmer said. “It was an anomaly but we could thing in Vermont because we’ve kept money get to 200 this year.” out of the equation.” In addition to its use on pancakes and As in years past, there is free admission to waffles, syrup can be used for all different rea- the Maple Festival. -

Review Article MRI Findings in Neuroferritinopathy
Hindawi Publishing Corporation Neurology Research International Volume 2012, Article ID 197438, 7 pages doi:10.1155/2012/197438 Review Article MRI Findings in Neuroferritinopathy Emiko Ohta and Yoshihisa Takiyama Department of Neurology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan Correspondence should be addressed to Emiko Ohta, [email protected] Received 12 March 2011; Revised 10 May 2011; Accepted 23 May 2011 Academic Editor: Antonio Cerasa Copyright © 2012 E. Ohta and Y. Takiyama. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Neuroferritinopathy is a neurodegenerative disease which demonstrates brain iron accumulation caused by the mutations in the ferritin light chain gene. On brain MRI in neuroferritinopathy, iron deposits are observed as low-intensity areas on T2WI and as signal loss on T2∗WI. On T2WI, hyperintense abnormalities reflecting tissue edema and gliosis are also seen. Another characteristic finding is the presence of symmetrical cystic changes in the basal ganglia, which are seen in the advanced stages of this disorder. Atrophy is sometimes noted in the cerebellar and cerebral cortices. The variety in the MRI findings is specific to neuroferritinopathy. Based on observations of an excessive iron content in patients with chronic neurologic disorders, such as Parkinson disease and Alzheimer disease, the presence of excess iron is therefore recognized as a major risk factor for neurodegenerative diseases. The future development of multimodal and advanced MRI techniques is thus expected to play an important role in accurately measuring the brain iron content and thereby further elucidating the neurodegenerative process. -

2006 Kyoto, Japan
October 28 - November 2, 2006 ~ Kyoto, Japan ~ Final Program Table of Contents Welcome . .2 Acknowledgements . .3 Organization . .6 MDS .Committees .& Task. .Forces . .9 International .Congress .Registration .and Venue. .12 International .Congress .Information . 13-15 . Continuing .Medical .Education . .13 . Evaluations . .14 . Press .Room . .15 Program-at-a-Glance . .17 Scientific Session Definitions . .19 Scientific .Sessions . .21 Faculty . .51 Committee .& Task. .Force .Meetings . .56 Exhibitor .Information . .57 Exhibitor .Directory . .58 Floor .Plans . 62-64 Map .of .Kyoto . .66 Lunch Map . .67 Subway Map . .68 Social Events . .69 Poster .Session .1 . .72 Poster .Session .2 . .88 Poster .Session .3 . .102 Poster .Session .4 . .117 CME .Request .Form . .133 The Movement. .Disorder .Society’s 0th International Congress of Parkinson’s Disease and Movement Disorders Welcome Letter Dear Colleagues, On behalf of The Movement Disorder Society (MDS), we are pleased to welcome you to Kyoto, Japan for the 10th International Congress of Parkinson’s Disease and Movement Disorders . The 10th International Congress has been designed to provide an innovative and comprehensive overview of the latest perspectives and research developments in the field of Movement Disorders . We encourage you to take every opportunity to participate in the Scientific Program which has drawn world renowned speakers and foremost experts in their respective fields . In the next days, the latest research regarding Movement Disorders will be presented and discussed in an open format, offering unique educational opportunities for all delegates . The International Congress convenes with a series of Opening Seminars and then continues with an array of Plenary, Parallel, Poster and Video Sessions, as well as Lunch Seminars, Controversies and Skills Workshops . -

Recommendations of the International Parkinson and Movement Disorder
Recommendations of the International Parkinson and Movement Disorder Society Task Force on Nomenclature of Genetic Movement Disorders Connie Marras 1 MD, PhD, Anthony Lang MD 1, Bart P. van de Warrenburg,4 Carolyn Sue, 3 Sarah J. Tabrizi MBChB, PhD, 5 Lars Bertram MD, 6,7 Katja Lohmann 2 PhD, Saadet Mercimek-Mahmutoglu, MD, PhD, 8 Alexandra Durr 9, Vladimir Kostic 10 , Christine Klein 2 MD, 1Toronto Western Hospital Morton and Gloria Shulman Movement Disorders Centre and the Edmond J. Safra Program in Parkinson’s disease, University of Toronto, Toronto, Canada 2Institute of Neurogenetics, University of Lübeck, Lübeck, Germany 3Department of Neurology, Royal North Shore Hospital and Kolling Institute of Medical Research, University of Sydney, St Leonards, NSW 2065, Australia 4Department of Neurology, Donders Institute for Brain, Cognition, and Behaviour, Radboud University Medical Centre, Nijmegen, The Netherlands 5Department of Neurodegenerative Disease, Institute of Neurology, University College London, UK 6 Platform for Genome Analytics, Institute of Neurogenetics, University of Lübeck, Lübeck, Germany 7School of Public Health, Faculty of Medicine, Imperial College, London, UK 8Division of Clinical and Metabolic Genetics, Department of Pediatrics, University of Toronto, The Hospital for Sick Children, Toronto, Canada 9 Sorbonne Université, UPMC Univ Paris 06, UM 75, ICM, F-75013 Paris, France; Inserm, U 1127, ICM, F-75013 Paris, France; Cnrs, UMR 7225, ICM, F-75013 Paris, France; ICM, Paris, F-75013 Paris, France; AP-HP, Hôpital de -

Iain Sinclair
Iain Sinclair: An Inventory of His Papers at the Harry Ransom Center Descriptive Summary Creator: Sinclair, Iain, 1943- Title: Iain Sinclair Papers Dates: 1882-2009 (bulk 1960s-2008) Extent: 135 document boxes, 8 oversize boxes (osb) (56.7 linear feet), 23 oversize folders (osf), 15 computer disks Abstract: The papers of British writer Iain Sinclair consist of drafts of works, research material, juvenilia, notebooks, personal and professional correspondence, business files, financial files, works by others, ephemera, and electronic files. They document Sinclair’s prolific and diverse career, from running his own press to his wide range of creative output including works of poetry, fiction, non-fiction, edited anthologies, screenplays, articles, essays, reviews, and radio and television contributions. Call Number: Manuscript Collection MS-4930 Language: English; some French, German, and Italian Access: Open for research. Researchers must register and agree to copyright and privacy laws before using archival materials. Some materials restricted due to condition and conservation status. Use Policies: Ransom Center collections may contain material with sensitive or confidential information that is protected under federal or state right to privacy laws and regulations. Researchers are advised that the disclosure of certain information pertaining to identifiable living individuals represented in the collections without the consent of those individuals may have legal ramifications (e.g., a cause of action under common law for invasion of privacy may arise if facts concerning an individual's private life are published that would be deemed highly offensive to a reasonable person) for which the Ransom Center and The University of Texas at Austin assume no responsibility. -

ASSESSED VALUE REVISIONS Downers Grove Township, 2019
NOTICE TO TAXPAYERS – ASSESSED VALUE REVISIONS Downers Grove Township, 2019 Assessed Values Median Level of Assessment: 33 1/3% Valuation Date: 01/01/2019 Relevant Real Estate Sales Information Used to Develop the Equalized Assessed Value: Sales Occurring Between 01/01/2016 – 12/31/2018 Your property is to be assessed at the above listed median level of assessment for the assessment district. You may check the accuracy of your assessment by dividing your assessment by the median level of assessment. The resulting value should equal the estimated fair cash value of your property. If the resulting value is greater than the estimated fair cash value of your property, you may be over-assessed. If the resulting value is less than the fair cash value of your property, you may be under-assessed. You may appeal your assessment to the Board of Review Illinois law requires assessed values of property, other than farmland and coal, are to be assessed of 33 1/3% of fair market value. Restrictions within Illinois law delay the change in property assessments caused by changes of actual property fair cash value. State law requires the assessed values are to be adjusted based upon data from the three prior calendar years before the assessment date. In appreciating markets, this forces current property assessments to lag behind recent sales prices, and in declining markets, the decline of assessed values is delayed. Contact your local assessor’s office to review your assessment if you believe the fair cash value is incorrect or you are not assessed uniformly with comparable properties in your neighborhood.