Diagnosis of Neurogenetic Disorders Contribution of Next Generation Sequencing and Deep Phenotyping
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Parkinson's Disease and Metal Storage Disorders
brain sciences Review Parkinson’s Disease and Metal Storage Disorders: A Systematic Review Edward Botsford 1,* , Jayan George 2,3 and Ellen E. Buckley 4,5 1 University of Sheffield Medical School, Beech Hill Road, Sheffield S10 2RX, UK 2 General Surgical Department, Sheffield Teaching Hospitals NHS Foundation Trust, Herries Road, Sheffield S5 7AU, UK; [email protected] 3 University of Sheffield, Western Bank, S10 2TN Sheffield, UK 4 Sheffield Institute for Translational Neuroscience, University of Sheffield, 385a Glossop Road, Sheffield S10 2HQ, UK; e.e.buckley@sheffield.ac.uk 5 INSIGNEO Institute for in silico Medicine, University of Sheffield, Pam Liversidge Building, Sheffield S1 3JD, UK * Correspondence: ebotsford1@sheffield.ac.uk; Tel.: +44-(0)114-222-2272 Received: 9 October 2018; Accepted: 30 October 2018; Published: 31 October 2018 Abstract: Metal storage disorders (MSDs) are a set of rare inherited conditions with variable clinical pictures including neurological dysfunction. The objective of this study was, through a systematic review, to identify the prevalence of Parkinsonism in patients with MSDs in order to uncover novel pathways implemented in Parkinson’s disease. Human studies describing patients of any age with an MSD diagnosis were analysed. Foreign language publications as well as animal and cellular studies were excluded. Searches were conducted through PubMed and Ovid between April and September 2018. A total of 53 publications were identified including 43 case reports, nine cross-sectional studies, and one cohort study. The publication year ranged from 1981 to 2018. The most frequently identified MSDs were Pantothenate kinase-associated neurodegeneration (PKAN) with 11 papers describing Parkinsonism, Hereditary hemochromatosis (HH) (7 papers), and Wilson’s disease (6 papers). -

A Decision Support System for Reporting High Throughput Sequencing of Cancers in Clinical Diagnostic Laboratories Kenneth D
Doig et al. Genome Medicine (2017) 9:38 DOI 10.1186/s13073-017-0427-z SOFTWARE Open Access PathOS: a decision support system for reporting high throughput sequencing of cancers in clinical diagnostic laboratories Kenneth D. Doig1,2,3,8*, Andrew Fellowes2, Anthony H. Bell2, Andrei Seleznev2, David Ma2, Jason Ellul1, Jason Li1, Maria A. Doyle1, Ella R. Thompson2,3, Amit Kumar1,6,7, Luis Lara1,3, Ravikiran Vedururu2, Gareth Reid2, Thomas Conway1, Anthony T. Papenfuss1,3,5,6 and Stephen B. Fox2,3,4 Abstract Background: The increasing affordability of DNA sequencing has allowed it to be widely deployed in pathology laboratories. However, this has exposed many issues with the analysis and reporting of variants for clinical diagnostic use. Implementing a high-throughput sequencing (NGS) clinical reporting system requires a diverse combination of capabilities, statistical methods to identify variants, global variant databases, a validated bioinformatics pipeline, an auditable laboratory workflow, reproducible clinical assays and quality control monitoring throughout. These capabilities must be packaged in software that integrates the disparate components into a useable system. Results: To meet these needs, we developed a web-based application, PathOS, which takes variant data from a patient sample through to a clinical report. PathOS has been used operationally in the Peter MacCallum Cancer Centre for two years for the analysis, curation and reporting of genetic tests for cancer patients, as well as the curation of large-scale research studies. PathOS has also been deployed in cloud environments allowing multiple institutions to use separate, secure and customisable instances of the system. Increasingly, the bottleneck of variant curation is limiting the adoption of clinical sequencing for molecular diagnostics. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetic Testing for Familial Alzheimer's Disease
Corporate Medical Policy Genetic Testing for Familial Alzheimer’s Disease AHS – M2038 File Name: genetic_testing_for_familial_alzheimers_disease Origination: 1/2019 Last CAP Review: 10/2020 Next CAP Review: 10/2021 Last Review: 10/2020 Description of Procedure or Service Alzheimer disease (AD) is a neurodegenerative disease defined by a gradual decline in memory, cognitive functions, gross atrophy of the brain, and an accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles (Karch, Cruchaga, & Goate, 2014). Familial Alzheimer disease (FAD) is a rare, inherited form of AD. FAD has a much earlier onset than other forms of Alzheimer disease with symptoms developing in individuals in their thirties or forties. Related Policies General Genetic Testing, Germline Disorders AHS – M2145 General Genetic Testing, Somatic Disorders AHS – M2146 ***Note: This Medical Policy is complex and technical. For questions concerning the technical language and/or specific clinical indications for its use, please consult your physician. Policy BCBSNC will provide coverage for genetic testing for familial Alzheimer disease when it is determined the medical criteria or reimbursement guidelines below are met. Benefits Application This medical policy relates only to the services or supplies described herein. Please refer to the Member's Benefit Booklet for availability of benefits. Member's benefits may vary according to benefit design; therefore member benefit language should be reviewed before applying the terms of this medical policy. -

A Pilot Trial of Deferiprone for Neurodegeneration with Brain Iron Accumulation
BRIEF REPORTS A pilot trial of deferiprone for neurodegeneration with brain iron accumulation Giovanni Abbruzzese, 1 Giovanni Cossu, 2 Manuela Balocco, 3 Roberta Marchese, 1 Daniela Murgia, 2 Maurizio Melis, 2 Renzo Galanello, 4 Susanna Barella, 4 Gildo Matta, 5 Uberto Ruffinengo, 6 Ubaldo Bonuccelli, 7 and Gian Luca Forni, 3 1Department of Neurosciences, University of Genoa; 2Neurology Department, “G. Brotzu” General Hospital, Cagliari; 3Centro della Microcitemia e Anemie Congenite-Haematology, Galliera Hospital, Genoa; 4Ospedale Regionale Microcitemia, University of Cagliari, Cagliari; 5Radiology Department “G. Brotzu” General Hospital, Cagliari; 6Neuroradiology Unit, Galliera Hospital, Genoa; 7Neuroscience Department, University of Pisa, Italy ABSTRACT Deferiprone was shown to reverse iron deposition in associated neurodegeneration). These results underline the Friedreich's ataxia. This multi-center, unblinded, single-arm safety and tolerability of deferiprone, and suggest that pilot study evaluated safety and efficacy of deferiprone for chelating treatment might be effective in improving neuro - reducing cerebral iron accumulation in neurodegeneration logical manifestations associated with iron accumulation. with brain iron accumulation. Four patients with genetical - (Clinicaltrials.gov Identifier: NTC00907283) ly-confirmed pantothenate kinase-associated neurodegener - ation, and 2 with parkinsonism and focal dystonia, but Key words: deferiprone, iron overload, neurodegeneration inconclusive genetic tests, received 15 mg/kg deferiprone with brain iron accumulation. bid. Magnetic resonance imaging and neurological examina - tions were conducted at baseline, six and 12 months. Citation: Abbruzzese G, Cossu G, Balocco M, Marchese R, Chelation treatment caused no apparent hematologic or Murgia D, Melis M, Galanello R, Barella S, Matta G, neurological side effects. Magnetic resonance imaging Ruffinengo U, Bonuccelli U, and Forni GL. -

Locus Reference Genomic Sequences: an Improved Basis for Describing Human DNA Variants
Dalgleish et al. Genome Medicine 2010, 2:24 http://genomemedicine.com/content/2/4/24 CORRESPONDENCE Open Access Locus Reference Genomic sequences: an improved basis for describing human DNA variants Raymond Dalgleish1*, Paul Flicek 2, Fiona Cunningham2, Alex Astashyn3, Raymond E Tully3, Glenn Proctor2, Yuan Chen2, William M McLaren2, Pontus Larsson2, Brendan W Vaughan2, Christophe Béroud4, Glen Dobson5, Heikki Lehväslaiho6, Peter EM Taschner7, Johan T den Dunnen7, Andrew Devereau5, Ewan Birney2, Anthony J Brookes1 and Donna R Maglott3 Introduction Abstract In 1993 Ernest Beutler wrote an eloquent letter to the As our knowledge of the complexity of gene editor of the American Journal of Human Genetics high- architecture grows, and we increase our understanding lighting the defi ciencies of the systems then used to of the subtleties of gene expression, the process of describe DNA variants [1]. Th at same year, the editor of accurately describing disease-causing gene variants Human Mutation invited Arthur Beaudet and Lap-Chee has become increasingly problematic. In part, this is Tsui to produce a nomenclature for variants in genes and due to current reference DNA sequence formats that proteins [2]. From these simple beginnings, the last 17 do not fully meet present needs. Here we present the years have borne witness to the steady development of Locus Reference Genomic (LRG) sequence format, the nomenclature used to describe sequence variation which has been designed for the specifi c purpose that is now maintained under the auspices of the Human of gene variant reporting. The format builds on Genome Variation Society (HGVS) [3,4]. To some, the the successful National Center for Biotechnology present nomenclature may seem like an arcane art-form Information (NCBI) RefSeqGene project and provides jealously guarded by zealots. -

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

Insurance and Advance Pay Test Requisition
Insurance and Advance Pay Test Requisition (2021) For Specimen Collection Service, Please Fax this Test Requisition to 1.610.271.6085 Client Services is available Monday through Friday from 8:30 AM to 9:00 PM EST at 1.800.394.4493, option 2 Patient Information Patient Name Patient ID# (if available) Date of Birth Sex designated at birth: 9 Male 9 Female Street address City, State, Zip Mobile phone #1 Other Phone #2 Patient email Language spoken if other than English Test and Specimen Information Consult test list for test code and name Test Code: Test Name: Test Code: Test Name: 9 Check if more than 2 tests are ordered. Additional tests should be checked off within the test list ICD-10 Codes (required for billing insurance): Clinical diagnosis: Age at Initial Presentation: Ancestral Background (check all that apply): 9 African 9 Asian: East 9 Asian: Southeast 9 Central/South American 9 Hispanic 9 Native American 9 Ashkenazi Jewish 9 Asian: Indian 9 Caribbean 9 European 9 Middle Eastern 9 Pacific Islander Other: Indications for genetic testing (please check one): 9 Diagnostic (symptomatic) 9 Predictive (asymptomatic) 9 Prenatal* 9 Carrier 9 Family testing/single site Relationship to Proband: If performed at Athena, provide relative’s accession # . If performed at another lab, a copy of the relative’s report is required. Please attach detailed medical records and family history information Specimen Type: Date sample obtained: __________ /__________ /__________ 9 Whole Blood 9 Serum 9 CSF 9 Muscle 9 CVS: Cultured 9 Amniotic Fluid: Cultured 9 Saliva (Not available for all tests) 9 DNA** - tissue source: Concentration ug/ml Was DNA extracted at a CLIA-certified laboratory or a laboratory meeting equivalent requirements (as determined by CAP and/or CMS)? 9 Yes 9 No 9 Other*: If not collected same day as shipped, how was sample stored? 9 Room temp 9 Refrigerated 9 Frozen (-20) 9 Frozen (-80) History of blood transfusion? 9 Yes 9 No Most recent transfusion: __________ /__________ /__________ *Please contact us at 1.800.394.4493, option 2 prior to sending specimens. -

Eutherian Adaptation from a TRAK-Like Protein, Conserved Gene Promoter Elements, and Localization in the Human Intestine Amanda L
Lumsden et al. BMC Evolutionary Biology (2016) 16:214 DOI 10.1186/s12862-016-0780-3 RESEARCH ARTICLE Open Access Huntingtin-associated protein 1: Eutherian adaptation from a TRAK-like protein, conserved gene promoter elements, and localization in the human intestine Amanda L. Lumsden1*, Richard L. Young2,3, Nektaria Pezos2,3 and Damien J. Keating1,2* Abstract Background: Huntingtin-associated Protein 1 (HAP1) is expressed in neurons and endocrine cells, and is critical for postnatal survival in mice. HAP1 shares a conserved “HAP1_N” domain with TRAfficking Kinesin proteins TRAK1 and TRAK2 (vertebrate), Milton (Drosophila) and T27A3.1 (C. elegans). HAP1, TRAK1 and TRAK2 have a degree of common function, particularly regarding intracellular receptor trafficking. However, TRAK1, TRAK2 and Milton (which have a “Milt/TRAK” domain that is absent in human and rodent HAP1) differ in function to HAP1 in that they are mitochondrial transport proteins, while HAP1 has emerging roles in starvation response. We have investigated HAP1 function by examining its evolution, and upstream gene promoter sequences. We performed phylogenetic analyses of the HAP1_N domain family of proteins, incorporating HAP1 orthologues (identified by genomic synteny) from 5 vertebrate classes, and also searched the Dictyostelium proteome for a common ancestor. Computational analyses of mammalian HAP1 gene promoters were performed to identify phylogenetically conserved regulatory motifs. Results: We found that as recently as marsupials, HAP1 contained a Milt/TRAK domain and was more similar to TRAK1 and TRAK2 than to eutherian HAP1. The Milt/TRAK domain likely arose post multicellularity, as it was absent in the Dictyostelium proteome. It was lost from HAP1 in the eutherian lineage, and also from T27A3.1 in C. -

Review Article MRI Findings in Neuroferritinopathy
Hindawi Publishing Corporation Neurology Research International Volume 2012, Article ID 197438, 7 pages doi:10.1155/2012/197438 Review Article MRI Findings in Neuroferritinopathy Emiko Ohta and Yoshihisa Takiyama Department of Neurology, Interdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan Correspondence should be addressed to Emiko Ohta, [email protected] Received 12 March 2011; Revised 10 May 2011; Accepted 23 May 2011 Academic Editor: Antonio Cerasa Copyright © 2012 E. Ohta and Y. Takiyama. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Neuroferritinopathy is a neurodegenerative disease which demonstrates brain iron accumulation caused by the mutations in the ferritin light chain gene. On brain MRI in neuroferritinopathy, iron deposits are observed as low-intensity areas on T2WI and as signal loss on T2∗WI. On T2WI, hyperintense abnormalities reflecting tissue edema and gliosis are also seen. Another characteristic finding is the presence of symmetrical cystic changes in the basal ganglia, which are seen in the advanced stages of this disorder. Atrophy is sometimes noted in the cerebellar and cerebral cortices. The variety in the MRI findings is specific to neuroferritinopathy. Based on observations of an excessive iron content in patients with chronic neurologic disorders, such as Parkinson disease and Alzheimer disease, the presence of excess iron is therefore recognized as a major risk factor for neurodegenerative diseases. The future development of multimodal and advanced MRI techniques is thus expected to play an important role in accurately measuring the brain iron content and thereby further elucidating the neurodegenerative process. -

Recommendations of the International Parkinson and Movement Disorder
Recommendations of the International Parkinson and Movement Disorder Society Task Force on Nomenclature of Genetic Movement Disorders Connie Marras 1 MD, PhD, Anthony Lang MD 1, Bart P. van de Warrenburg,4 Carolyn Sue, 3 Sarah J. Tabrizi MBChB, PhD, 5 Lars Bertram MD, 6,7 Katja Lohmann 2 PhD, Saadet Mercimek-Mahmutoglu, MD, PhD, 8 Alexandra Durr 9, Vladimir Kostic 10 , Christine Klein 2 MD, 1Toronto Western Hospital Morton and Gloria Shulman Movement Disorders Centre and the Edmond J. Safra Program in Parkinson’s disease, University of Toronto, Toronto, Canada 2Institute of Neurogenetics, University of Lübeck, Lübeck, Germany 3Department of Neurology, Royal North Shore Hospital and Kolling Institute of Medical Research, University of Sydney, St Leonards, NSW 2065, Australia 4Department of Neurology, Donders Institute for Brain, Cognition, and Behaviour, Radboud University Medical Centre, Nijmegen, The Netherlands 5Department of Neurodegenerative Disease, Institute of Neurology, University College London, UK 6 Platform for Genome Analytics, Institute of Neurogenetics, University of Lübeck, Lübeck, Germany 7School of Public Health, Faculty of Medicine, Imperial College, London, UK 8Division of Clinical and Metabolic Genetics, Department of Pediatrics, University of Toronto, The Hospital for Sick Children, Toronto, Canada 9 Sorbonne Université, UPMC Univ Paris 06, UM 75, ICM, F-75013 Paris, France; Inserm, U 1127, ICM, F-75013 Paris, France; Cnrs, UMR 7225, ICM, F-75013 Paris, France; ICM, Paris, F-75013 Paris, France; AP-HP, Hôpital de -
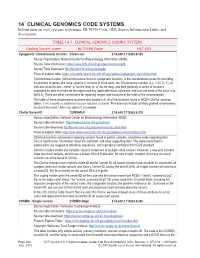
14 CLINICAL GENOMICS CODE SYSTEMS Information on Code Systems with Name, HL70396 Code, OID, Source Information Links, and Description
14 CLINICAL GENOMICS CODE SYSTEMS Information on code systems with name, HL70396 Code, OID, Source Information Links, and description. TABLE 14-1. CLINICAL GENOMICS CODING SYSTEMS Coding System name HL70396 Code HL7 OID Cytogenetic (chromosome) location Chrom-Loc 2.16.840.1.113883.6.335 Source Organization: National Center for Biotechnology Information (NCBI) Source Table Information: https://www.ncbi.nlm.nih.gov/genome/tools/gdp Source Table Download: ftp://ftp.ncbi.nlm.nih.gov/pub/gdp Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/cytogenetic_locs/v3/doc.html Chromosome location (AKA chromosome locus or cytogenetic location), is the standardized syntax for recording the position of genes and large variants. It consists of three parts: the Chromosome number (e.g. 1-22, X, Y), an indicator of which arm – either “p” for the short or “q” for the long, and then generally a series of numbers separated by dots that indicate the region and any applicable band, sub-band, and sub-sub-band of the locus (e.g. 2p16.3). There are other conventions for reporting ranges and locations at the ends of the chromosomes. The table of these chromosome locations was loaded with all of the locations found in NCBI’s ClinVar variation tables. It will expand as additional sources become available. This does not include all finely grained chromosome locations that exist. Users can add to it as needed. ClinVar Variant ID CLINVAR-V 2.16.840.1.113883.6.319 Source organization: National Center for Biotechnology Information (NCBI) Source table information: http://www.ncbi.nlm.nih.gov/clinvar/ Source table download: ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/tab_delimited/ Place to explore table: https://clin-table-search.lhc.nlm.nih.gov/apidoc/variants/v3/doc.html ClinVar processes submissions reporting variants found in patient samples, assertions made regarding their clinical significance, information about the submitter, and other supporting data.