Functional Characterization of Novel Rhot1 Variants, Which Are Associated with Parkinson’S Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
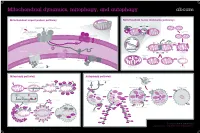
Mitochondrial Dynamics, Mitophagy, and Autophagy
Mitochondrial dynamics, mitophagy, and autophagy Mitochondrial import protein pathway Mitochondrial fusion and fission pathways iΔΨm, (OXPHOSh) Ca2+ Fusion ++++ (Cell differentiationi) N Internal signal β-barrel outer Opa1 SH ER sequence SH membrane precursors Mfn1/2 SH SH GTP GTP C Inner membrane and GTP GTP Outer membrane Matrix carrier precursors 22 70 GTP fusion (N-terminal presequences) 35 GTP 20 37 Tom complex 5 Sam complex Opa1 Cytosol Outer 6 Sam50 7 Mdm10 membrane DRP1 Fis1 Inner membrane fusion Tim8-Tim13 Mim 1 Tom40 Bax/Bak apoptosis Outer membrane HR2 regions S-S Tim9-Tim10 S-S Inner Erv1 95 A 54 mitochondrial S-S Reorganization Mia40 space 21 Fission sequestration 50 18 Tim22 Translocation Depolarization to meet iΔΨm ATP needs Tim23 KREBS Cycle NADH FADH2 17 Oxa1 Tim 22 complex Mia Mba1 Mdm I ATP 44 38 H+ II 17 H+ Inner membrane 16 +++ III ADP - Pi 18 Ribosome Oxa H+ IV mtHsp70 H+ ATP +++ Mge1 MPP Healthy mitochondrion Mitophagy Matrix Tim 23 complex Mitophagy pathway Autophagy pathway Lysosomal hydroiase Lysosome Hypoxia, Nutrient/Growth Rapamycin Starvation condition Ubiquitin Cytosol Parkin factor deprivation Lamp Stress Damaged mitochondrion Parkin Parkin iΔΨm Parkin MARF PINK1 PINK1 PINK1 Mfn1 VDAC1 Parkin AMPK mTOR Mfn2 PINK1 PINK1 Phagophore Autophagosome Autophagolyosome PARL LC3-II PINK1-L PINK1-S Parkin P FIS1 ATG13 ULK LC3-II Membrane PINK1 PINK1 FIP200 PINK1 MIRO2 P Bak PINK1 MIRO1 Parkin Parkin Parkin Cytoplasmic Cytosol proLC3 macromolecule LC3-II Atg4B Atg7/LC3-I Atg5/Atg12/Atg16 Organelle Atg9 PE Atg18 Atg2 -

ARMCX3 (NM 177948) Human Untagged Clone – SC124834
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for SC124834 ARMCX3 (NM_177948) Human Untagged Clone Product data: Product Type: Expression Plasmids Product Name: ARMCX3 (NM_177948) Human Untagged Clone Tag: Tag Free Symbol: ARMCX3 Synonyms: ALEX3; dJ545K15.2; GASP6 Vector: pCMV6-XL4 E. coli Selection: Ampicillin (100 ug/mL) Cell Selection: None Fully Sequenced ORF: >NCBI ORF sequence for NM_177948, the custom clone sequence may differ by one or more nucleotides ATGGGCTACGCCAGGAAAGTAGGCTGGGTGACCGCAGGCCTGGTGATTGGGGCTGGCGCCTGCTATTGCA TTTATAGACTGACTAGGGGAAGAAAACAGAACAAGGAAAAAATGGCTGAGGGTGGATCTGGGGATGTGGA TGATGCTGGGGACTGTTCTGGGGCCAGGTATAATGACTGGTCTGATGATGATGATGACAGCAATGAGAGC AAGAGTATAGTATGGTACCCACCTTGGGCTCGGATTGGGACTGAAGCTGGAACCAGAGCTAGGGCCAGGG CAAGGGCCAGGGCTACCCGGGCACGTCGGGCTGTCCAGAAACGGGCTTCCCCCAATTCAGATGATACCGT TTTGTCCCCTCAAGAGCTACAAAAGGTTCTTTGCTTGGTTGAGATGTCTGAAAAGCCTTATATTCTTGAA GCAGCTTTAATTGCTCTGGGTAACAATGCTGCTTATGCATTTAACAGAGATATTATTCGTGATCTGGGTG GTCTCCCAATTGTCGCAAAGATTCTCAATACTCGGGATCCCATAGTTAAGGAAAAGGCTTTAATTGTCCT GAATAACTTGAGTGTGAATGCTGAAAATCAGCGCAGGCTTAAAGTATACATGAATCAAGTGTGTGATGAC ACAATCACTTCTCGCTTGAACTCATCTGTGCAGCTTGCTGGACTGAGATTGCTTACAAATATGACTGTTA CTAATGAGTATCAGCACATGCTTGCTAATTCCATTTCTGACTTTTTTCGTTTATTTTCAGCGGGAAATGA AGAAACCAAACTTCAGGTTCTGAAACTCCTTTTGAATTTGGCTGAAAATCCAGCCATGACTAGGGAACTG CTCAGGGCCCAAGTACCATCTTCACTGGGCTCCCTCTTTAATAAGAAGGAGAACAAAGAAGTTATTCTTA AACTTCTGGTCATATTTGAGAACATAAATGATAATTTCAAATGGGAAGAAAATGAACCTACTCAGAATCA -

PARSANA-DISSERTATION-2020.Pdf
DECIPHERING TRANSCRIPTIONAL PATTERNS OF GENE REGULATION: A COMPUTATIONAL APPROACH by Princy Parsana A dissertation submitted to The Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July, 2020 © 2020 Princy Parsana All rights reserved Abstract With rapid advancements in sequencing technology, we now have the ability to sequence the entire human genome, and to quantify expression of tens of thousands of genes from hundreds of individuals. This provides an extraordinary opportunity to learn phenotype relevant genomic patterns that can improve our understanding of molecular and cellular processes underlying a trait. The high dimensional nature of genomic data presents a range of computational and statistical challenges. This dissertation presents a compilation of projects that were driven by the motivation to efficiently capture gene regulatory patterns in the human transcriptome, while addressing statistical and computational challenges that accompany this data. We attempt to address two major difficulties in this domain: a) artifacts and noise in transcriptomic data, andb) limited statistical power. First, we present our work on investigating the effect of artifactual variation in gene expression data and its impact on trans-eQTL discovery. Here we performed an in-depth analysis of diverse pre-recorded covariates and latent confounders to understand their contribution to heterogeneity in gene expression measurements. Next, we discovered 673 trans-eQTLs across 16 human tissues using v6 data from the Genotype Tissue Expression (GTEx) project. Finally, we characterized two trait-associated trans-eQTLs; one in Skeletal Muscle and another in Thyroid. Second, we present a principal component based residualization method to correct gene expression measurements prior to reconstruction of co-expression networks. -

Amyloid Β-Peptide Increases Mitochondria-Endoplasmic
cells Article Amyloid β-Peptide Increases Mitochondria-Endoplasmic Reticulum Contact Altering Mitochondrial Function and Autophagosome Formation in Alzheimer’s Disease-Related Models Nuno Santos Leal 1,*, Giacomo Dentoni 1 , Bernadette Schreiner 1 , Luana Naia 1, Antonio Piras 1, Caroline Graff 1, Antonio Cattaneo 2, Giovanni Meli 2 , Maho Hamasaki 3, Per Nilsson 1 and Maria Ankarcrona 1,* 1 Division of Neurogeriatrics, Department of Neurobiology, Care Science and Society, Karolinska Institutet, BioClinicum J9:20, Visionsgatan 4, 171 64 Solna, Sweden; [email protected] (G.D.); [email protected] (B.S.); [email protected] (L.N.); [email protected] (A.P.); caroline.graff@ki.se (C.G.); [email protected] (P.N.) 2 European Brain Research Institute (EBRI), Viale Regina Elena 295, 00161 Roma, Italy; [email protected] (A.C.); [email protected] (G.M.) 3 Department of Genetics, Graduate School of Medicine, Osaka University, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan; [email protected] * Correspondence: [email protected] (N.S.L.); [email protected] (M.A.); Tel.: +44-122-333-4390 (N.S.L.); +46-852-483-577 (M.A.) Received: 23 October 2020; Accepted: 25 November 2020; Published: 28 November 2020 Abstract: Recent findings have shown that the connectivity and crosstalk between mitochondria and the endoplasmic reticulum (ER) at mitochondria–ER contact sites (MERCS) are altered in Alzheimer’s disease (AD) and in AD-related models. MERCS have been related to the initial steps of autophagosome formation as well as regulation of mitochondrial function. Here, the interplay between MERCS, mitochondria ultrastructure and function and autophagy were evaluated in different AD animal models with increased levels of Aβ as well as in primary neurons derived from these animals. -

Involvement of Impaired Autophagy and Mitophagy in Neuro-2A Cell
www.nature.com/scientificreports OPEN Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high- Received: 19 May 2017 Accepted: 15 January 2018 glucose conditions Published: xx xx xxxx Yufei Song, Yu Du, Wenying Zou, Yan Luo, Xiaojie Zhang & Jianliang Fu Chronic cerebral hypoperfusion (CCH) plays an insidious role in the development of cognitive impairment. Considerable evidence suggests that Diabetes Mellitus (DM) as a vascular risk factor may exacerbate CCH and is closely related to cognitive decline. Dysregulation of autophagy is known to be associated with the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease. To elucidate the role of autophagy in CCH- and/or DM-related pathogenesis, mouse neuroblastoma Neuro-2a cells were exposed to hypoxia and/or high glucose for 48 h, mimicking CCH complicated with DM pathologies. Chronic hypoxia reduced cell proliferation and increased levels of cleaved caspase-3, whereas high glucose had no obvious synergistic toxic efect. Accumulation of autophagic vacuoles under hypoxia may be due to both autophagy impairment and induction, with the former accounting for Neuro-2a cell death. Additionally, aberrant accumulation of mitochondria in Neuro-2a cells may be attributed to insufcient BNIP3-mediated mitophagy due to poor interaction between BNIP3 and LC3-II. Despite the lack of a signifcant cytotoxic efect of high glucose under our experimental conditions, our data indicated for the frst time that impaired autophagy degradation and inefcient BNIP3-mediated mitophagy may constitute mechanisms underlying neuronal cell damage during chronic hypoxia. Chronic cerebral hypoperfusion (CCH) is a normal process related to ageing that likely contributes to age-related memory loss1. -

The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease
International Journal of Molecular Sciences Review The Mitochondrial Kinase PINK1 in Diabetic Kidney Disease Chunling Huang * , Ji Bian , Qinghua Cao, Xin-Ming Chen and Carol A. Pollock * Kolling Institute, Sydney Medical School, Royal North Shore Hospital, University of Sydney, St. Leonards, NSW 2065, Australia; [email protected] (J.B.); [email protected] (Q.C.); [email protected] (X.-M.C.) * Correspondence: [email protected] (C.H.); [email protected] (C.A.P.); Tel.: +61-2-9926-4784 (C.H.); +61-2-9926-4652 (C.A.P.) Abstract: Mitochondria are critical organelles that play a key role in cellular metabolism, survival, and homeostasis. Mitochondrial dysfunction has been implicated in the pathogenesis of diabetic kidney disease. The function of mitochondria is critically regulated by several mitochondrial protein kinases, including the phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1). The focus of PINK1 research has been centered on neuronal diseases. Recent studies have revealed a close link between PINK1 and many other diseases including kidney diseases. This review will provide a concise summary of PINK1 and its regulation of mitochondrial function in health and disease. The physiological role of PINK1 in the major cells involved in diabetic kidney disease including proximal tubular cells and podocytes will also be summarized. Collectively, these studies suggested that targeting PINK1 may offer a promising alternative for the treatment of diabetic kidney disease. Keywords: PINK1; diabetic kidney disease; mitochondria; mitochondria quality control; mitophagy Citation: Huang, C.; Bian, J.; Cao, Q.; 1. Introduction Chen, X.-M.; Pollock, C.A. -

PRODUCT SPECIFICATION Product Datasheet
Product Datasheet QPrEST PRODUCT SPECIFICATION Product Name QPrEST NRK Mass Spectrometry Protein Standard Product Number QPrEST30079 Protein Name Nik-related protein kinase Uniprot ID Q7Z2Y5 Gene NRK Product Description Stable isotope-labeled standard for absolute protein quantification of Nik-related protein kinase. Lys (13C and 15N) and Arg (13C and 15N) metabolically labeled recombinant human protein fragment. Application Absolute protein quantification using mass spectrometry Sequence (excluding GRRSQSSPPYSTIDQKLLVDIHVPDGFKVGKISPPVYLTNEWVGYNALSE fusion tag) IFRNDWLTPAPVIQPPEEDGDYVELYDASADTDGDDDDESNDTFEDTYDH ANGNDDLDNQVDQANDVCKD Theoretical MW 31219 Da including N-terminal His6ABP fusion tag Fusion Tag A purification and quantification tag (QTag) consisting of a hexahistidine sequence followed by an Albumin Binding Protein (ABP) domain derived from Streptococcal Protein G. Expression Host Escherichia coli LysA ArgA BL21(DE3) Purification IMAC purification Purity >90% as determined by Bioanalyzer Protein 230 Purity Assay Isotopic Incorporation >99% Concentration >5 μM after reconstitution in 100 μl H20 Concentration Concentration determined by LC-MS/MS using a highly pure amino acid analyzed internal Determination reference (QTag), CV ≤10%. Amount >0.5 nmol per vial, two vials supplied. Formulation Lyophilized in 100 mM Tris-HCl 5% Trehalose, pH 8.0 Instructions for Spin vial before opening. Add 100 μL ultrapure H2O to the vial. Vortex thoroughly and spin Reconstitution down. For further dilution, see Application Protocol. Shipping Shipped at ambient temperature Storage Lyophilized product shall be stored at -20°C. See COA for expiry date. Reconstituted product can be stored at -20°C for up to 4 weeks. Avoid repeated freeze-thaw cycles. Notes For research use only Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Inactive X Chromosome Is Epigenetically Unstable and Transcriptionally Labile in Breast Cancer
Supplemental Information The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer Ronan Chaligné1,2,3,8, Tatiana Popova1,4, Marco-Antonio Mendoza-Parra5, Mohamed-Ashick M. Saleem5 , David Gentien1,6, Kristen Ban1,2,3,8, Tristan Piolot1,7, Olivier Leroy1,7, Odette Mariani6, Hinrich Gronemeyer*5, Anne Vincent-Salomon*1,4,6,8, Marc-Henri Stern*1,4,6 and Edith Heard*1,2,3,8 Extended Experimental Procedures Cell Culture Human Mammary Epithelial Cells (HMEC, Invitrogen) were grown in serum-free medium (HuMEC, Invitrogen). WI- 38, ZR-75-1, SK-BR-3 and MDA-MB-436 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) containing 10% fetal bovine serum (FBS). DNA Methylation analysis. We bisulfite-treated 2 µg of genomic DNA using Epitect bisulfite kit (Qiagen). Bisulfite converted DNA was amplified with bisulfite primers listed in Table S3. All primers incorporated a T7 promoter tag, and PCR conditions are available upon request. We analyzed PCR products by MALDI-TOF mass spectrometry after in vitro transcription and specific cleavage (EpiTYPER by Sequenom®). For each amplicon, we analyzed two independent DNA samples and several CG sites in the CpG Island. Design of primers and selection of best promoter region to assess (approx. 500 bp) were done by a combination of UCSC Genome Browser (http://genome.ucsc.edu) and MethPrimer (http://www.urogene.org). All the primers used are listed (Table S3). NB: MAGEC2 CpG analysis have been done with a combination of two CpG island identified in the gene core. Analysis of RNA allelic expression profiles (based on Human SNP Array 6.0) DNA and RNA hybridizations were normalized by Genotyping console. -

Ageing-Associated Changes in DNA Methylation in X and Y Chromosomes
Kananen and Marttila Epigenetics & Chromatin (2021) 14:33 Epigenetics & Chromatin https://doi.org/10.1186/s13072-021-00407-6 RESEARCH Open Access Ageing-associated changes in DNA methylation in X and Y chromosomes Laura Kananen1,2,3,4* and Saara Marttila4,5* Abstract Background: Ageing displays clear sexual dimorphism, evident in both morbidity and mortality. Ageing is also asso- ciated with changes in DNA methylation, but very little focus has been on the sex chromosomes, potential biological contributors to the observed sexual dimorphism. Here, we sought to identify DNA methylation changes associated with ageing in the Y and X chromosomes, by utilizing datasets available in data repositories, comprising in total of 1240 males and 1191 females, aged 14–92 years. Results: In total, we identifed 46 age-associated CpG sites in the male Y, 1327 age-associated CpG sites in the male X, and 325 age-associated CpG sites in the female X. The X chromosomal age-associated CpGs showed signifcant overlap between females and males, with 122 CpGs identifed as age-associated in both sexes. Age-associated X chro- mosomal CpGs in both sexes were enriched in CpG islands and depleted from gene bodies and showed no strong trend towards hypermethylation nor hypomethylation. In contrast, the Y chromosomal age-associated CpGs were enriched in gene bodies, and showed a clear trend towards hypermethylation with age. Conclusions: Signifcant overlap in X chromosomal age-associated CpGs identifed in males and females and their shared features suggest that despite the uneven chromosomal dosage, diferences in ageing-associated DNA methylation changes in the X chromosome are unlikely to be a major contributor of sex dimorphism in ageing. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

PINK1, Parkin, and DJ-1 Mutations in Italian Patients with Early-Onset Parkinsonism
European Journal of Human Genetics (2005) 13, 1086–1093 & 2005 Nature Publishing Group All rights reserved 1018-4813/05 $30.00 www.nature.com/ejhg ARTICLE PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism Christine Klein*,1,2,9, Ana Djarmati1,2,3,9, Katja Hedrich1,2, Nora Scha¨fer1,2, Cesa Scaglione4, Roberta Marchese5, Norman Kock1,2, Birgitt Schu¨le1,2, Anja Hiller1, Thora Lohnau1,2, Susen Winkler1,2, Karin Wiegers1,2, Robert Hering6, Peter Bauer6, Olaf Riess6, Giovanni Abbruzzese5, Paolo Martinelli4 and Peter P Pramstaller7,8 1Department of Neurology, University of Lu¨beck, Lu¨beck, Germany; 2Department of Human Genetics, University of Lu¨beck, Lu¨beck, Germany; 3Faculty of Biology, University of Belgrade, Belgrade, Serbia; 4Institute of Neurology, University of Bologna, Bologna, Italy; 5Department of Neurology, University of Genova, Genova, Italy; 6Department of Medical Genetics, University of Tu¨bingen, Tu¨bingen, Germany; 7Department of Neurology, General Regional Hospital, Bolzano-Bozen, Italy; 8Department of Genetic Medicine, EURAC-Research, Bolzano-Bozen, Italy Recessively inherited early-onset parkinsonism (EOP) has been associated with mutations in the Parkin, DJ-1, and PINK1 genes. We studied the prevalence of mutations in all three genes in 65 Italian patients (mean age of onset: 43.275.4 years, 62 sporadic, three familial), selected by age at onset equal or younger than 51 years. Clinical features were compatible with idiopathic Parkinson’s disease in all cases. To detect small sequence alterations in Parkin, DJ-1, and PINK1, we performed a conventional mutational analysis (SSCP/ dHPLC/sequencing) of all coding exons of these genes.